
NH3对煤物理吸氧影响规律的量子化学研究*
2021-11-15 09:08张亚超郝朝瑜何文浩康延雷王三伟王雪峰
煤炭转化 2021年6期
张亚超 郝朝瑜 何文浩 康延雷 王三伟 王雪峰
(1.太原理工大学安全与应急管理工程学院,030024 太原;2.神华国能(神东电力)集团公司大南湖一矿,839112 新疆哈密)
0 引 言
煤炭作为世界各国的一种重要能源,其广泛应用于发电以及钢铁制造等多个行业,根据英国石油公司2019年《世界能源统计年鉴》的数据,2018年世界煤炭产量为39.17亿t,年增长4.3%[1-2]。煤炭作为发电的最大单一能源,占全球电力的36%以上,尤其在中国煤炭发电占比达到了72.83%[3]。在煤炭的开采以及利用过程中,煤的自燃、瓦斯爆炸灾害以及燃煤电厂烟气排放等问题一直是制约煤炭行业发展的主要问题[4-7]。煤与氧气相互作用发生氧化自燃反应的过程主要包括物理吸附、化学吸附和化学反应三个阶段,其中煤物理吸附氧气的阶段是引起煤与氧发生反应的开端。电厂烟气作为工业废弃物,氧含量极低,利用烟气注入井下惰化采空区能够起到预防煤自燃和瓦斯爆炸的作用,实现了防灾与减排的统一[8-12]。烟气普遍采用选择性催化还原以及非选择性催化还原(SCR/SNCR)技术进行脱硝,由于在脱硝过程中为获得更大的NOx去除效率[13-15],烟气中会存在部分“逃逸氨”[16-19],此外井下爆破、灭火及部分煤岩层中也含有氨气。目前,相关学者对于井下气体与煤分子的相互作用的研究主要集中在CH4,O2,CO2和H2O等气体[20-22],而对于氨气在煤层中的赋存状态以及其对采空区中烟气防灾封存效果的影响规律还不明确,需要进一步研究。
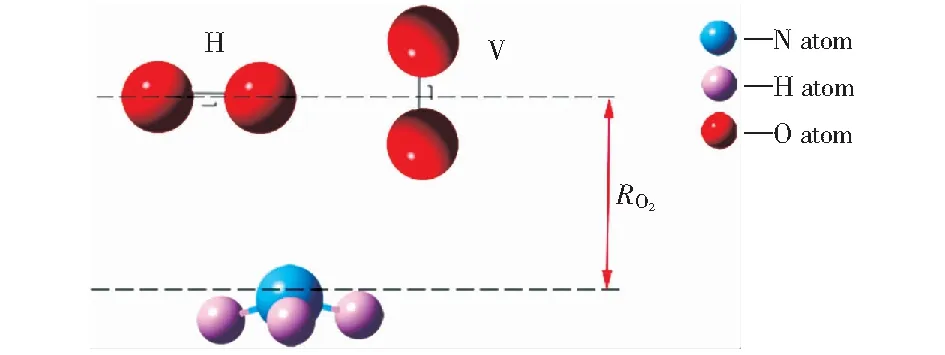
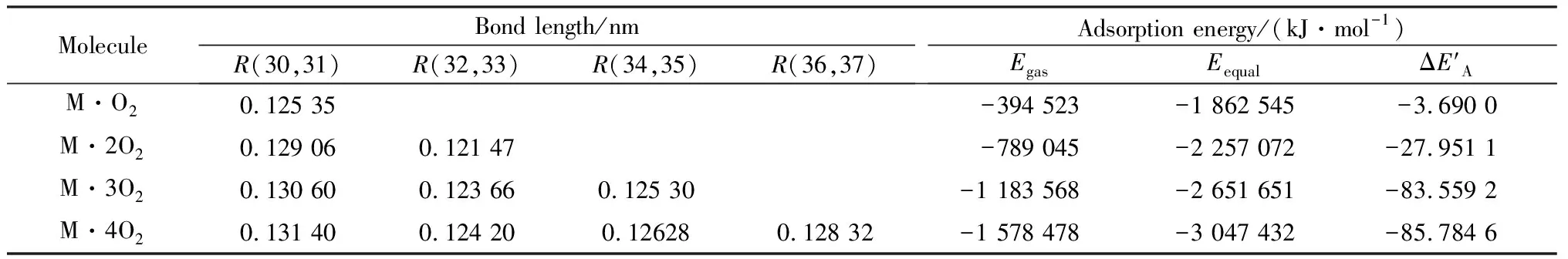
通过实验的方法对煤与不同气体的吸附以及不同气体在煤分子表面的相互作用得到了广泛研究[23-26]。然而,实验表征的方法只能从宏观角度对实验现象进行解释,很难对煤与不同气体分子以及不同气体分子之间的微观相互作用进行研究。近年来,随着计算化学的快速发展,借助理论模拟从分子水平对煤与气体之间相互作用的微观机理研究已大量出现[27-33]。王宝俊等[34]充分证明了量子化学方法在研究煤结构与反应性上的可行性,明确指出量子化学研究方法可以成功应用于煤的静态微观性质、吸附、解吸、裂解、液化和气化等研究中。邓存宝等[35-36]用量子化方法分别研究了含S,P—CH2CH3侧链基团的煤分子与O2之间的吸附作用,从理论模拟的水平上完善了煤自燃理论。LITHOXOOS et al[37]研究了N2,CH4,CO2在单壁碳纳米管上的吸附行为,得出它们在单壁碳纳米管上吸附能由大到小依次为:H2≤N2≈CH4 基于目前研究背景,本实验应用量子化学计算方法,从微观角度研究煤分子片段与NH3和O2分子的物理吸附过程,进而确定煤结构与NH3和O2之间的吸附模型,揭示它们彼此之间的吸附机理以及竞争吸附规律,研究矿井中NH3的存在对煤氧化自燃的影响,并对电厂烟气注入采空区之前的脱硝处理提出相关建议,以期进一步完善电厂烟气注入采空区防灭火技术,更好地发展烟气预防煤自燃理论。 由于煤大分子模型结构复杂,难以直接对其进行相关计算,当前对于煤分子结构的量子化学研究都需将煤大分子结构进行适度合理的简化。已有研究[38-39]表明,煤分子中芳香环的性质非常稳定,活性基团性质几乎不受煤分子模型影响,且煤分子中的烷基侧链主要为长链。因此,煤分子片段可以在最大程度保证煤大分子片段基础上简化为一个小分子片段,即煤的每个小分子只由一个活性基团、一个烷基侧链和芳香环组成。 煤基质中氮元素的质量分数很少(1%~2%),但煤基质表面含氮官能团对于煤体吸附性能却有显著影响,并且相关研究[40-41]表明虽然煤中的氨基侧链含量较少,但在煤氧化过程中,氨基侧链首先与氧发生物理吸附,进而发生化学反应放热,导致煤体温度升高,引起煤中其他类型的官能团氧化,并最终造成煤自燃。 山西作为煤炭生产基地,主要生产高变质程度烟煤与无烟煤,因此本次计算煤分子模型是基于高变质程度烟煤建立的。 基于上述结论以及煤结构“局部微观结构模型”基础性认识的观点,以王宝俊等[34]建立的低挥发分烟煤模型为基础对煤结构进行适度简化,使用GaussView 6.0软件构造出适合本研究的煤分子片段模型,该煤分子模型由两个苯环构成的煤分子骨架上伸出一条含有一个亚甲基(—CH2)和一个氨基(—NH2)相连构成的侧链和一条含有一个乙基(—C2H5)的侧链。在煤分子构建完成以后使用半经验方法(PM7)进行初步优化,将优化后的坐标输入Gaussian 16A,在B3LYP/6-311G(d,p)级别下进一步优化,得到优化后的结构如图1所示(其中RMS均方根)。经振动分析验证无虚频。 图1 煤分子优化过程的能量变化以及优化后的构型Fig.1 Energy change diagram of coal molecular optimization process and optimized configuration 对于煤这种有共轭体系的结构,共轭π电子的运动区域不再局限于两原子之间且离域现象比较明显,这时候电子相关就显得格外重要。本实验采用包含色散矫正的密度泛函(DFT-D3)方法对煤模型分子与NH3和O2进行吸附作用时的吸附过程进行相关计算。通过将NH3和O2分子以一定距离放置在煤分子表面的不同位置,在B3LYP-D3/6-311 G(d,p)级别下优化煤结构吸附气体构型,同时在相同计算水平对优化后的结构进行频率分析后检测到无虚频出现,验证优化后构型处于势能面的极小值点,接着调整NH3和O2放置在煤分子表面的距离以及位置,重复上述计算步骤,通过比较吸附体系的总能量,能量最低的构型即为能量最优吸附构型。接着在考虑基组重叠误差(basis set superpositionerror,BSSE)以及色散校正的情况下,基于M062X/def2-TZVP计算水平计算煤表面吸附气体分子的吸附能。吸附能的计算公式见式(1)和式(2): ΔE′=ΔE+EBSSE (1) ΔE=Eequal-Egas-Ecoal (2) 式中:ΔE′为考虑色散相互作用时的吸附能,kJ/mol;ΔE为未考虑BSSE矫正下的相互作用能,kJ/mol;EBSSE为色散作用能,kJ/mol;Eequal为煤与气体分子吸附作用过程中达到平衡时的吸附能,kJ/mol;Egas为气体分子发生吸附前的能量,kJ/mol;Ecoal为煤分子发生吸附前的能量,kJ/mol。由于吸附是一个放热过程,负值越低,吸附能越大,煤分子对气体的吸附能力越强。 对于两种气体在煤分子表面的共吸附体系,吸附能的计算公式见式(3)和式(4): (3) ΔEA=Ecoal+A+B-EA-Ecoal+B (4) 表1所示为在研究过程中使用到气体的相关参数,其值均为使用Gaussian 16A程序下优化后得到,其中气体分子的键长、键角、二面角以及振动频率的数值是基于B3LYP/6-311G(d,p)级别下优化计算得到,单点能的数值是将优化后的气体分子结构坐标作为新的高斯输入文件,使用M062X/def2-TZVP计算方法计算得到。 表1 气体相关参数Table 1 Relevant parameters of gases 静电势对于考察分子间静电相互作用、预测反应位点、预测分子性质等方面有重要意义。通过静电势我们可以方便、直观地了解分子与物质的相互作用,可以对结合强度、结合方式进行解释和预测[42-43]。通过将优化好的煤分子表面结构的波函数文件作为输入文件导入到Multiwfn程序中[44],计算分子表面的静电势,利用可视化程序VMD[45]将计算结果呈现出来,最终得到标有极值点的煤分子表面的静电势分布(如图2所示)。根据分子表面静电势计算结果,得到煤分子不同静电势区间分子表面积分布(如图3所示)。对于静电势区间的划分见图3中煤分子的原子序号所示,1号~16号原子为芳香环部分,17号~29号原子为非芳香环部分。 结合图2和图3可知,煤分子中的芳香环表面因具有丰富的π电子云使得其呈现出负静电势,且负静电势区域面积较大,占据了整个煤分子表面的36.93%,同时,由于芳香环中C的电负性大于H,使得H原子周围表现出正电性。由图3可以看出,非芳香环部分静电势(ESP)分布比芳香环部分ESP分布的覆盖范围更大且整个煤分子表面的静电势最大值以及最小值都位于非芳香环区域,这主要是极性较大的—NH2基团存在的原因。在非芳香环区域中,由于—NH2中的N原子电负性较强,会导致范德华表面的静电势最小值(最小值为-135.52 kJ/mol),而静电势最大值出现在—NH2中的H原子周围区域,最大值为+116.82 kJ/mol。 图2 煤分子表面静电势分布Fig.2 Electrostatic potential distribution of coal molecular surface(The deeper the blue is, the more negative the electrostatic potential is. The deeper the red is, the more positive the elec-trostatic potential is. The value of the electrostatic potential in the white area is around 0. The yellow ball is the maximum point of the surface electrostatic potential of coal molecules, and the cyan ball is the minimum point of the surface electro-static potential of coal molecules.) 图3 煤分子不同静电势区间分子表面积分布Fig.3 Surface area distribution of coal molecules in different electrostatic potential regions(The blue column is the molecular surface area distribution of different electrostatic potential regions on the surface of aro-matic ring, and the red column is the molecular surface area distribution of different electrostatic potential regions on the surface of non-aromatic ring. Ratio is the percentage of the total electrostatio potential region of different electrostatic potental regions on the molecular surface.) 图4所示为O2和NH3分子在—NH2侧链以及芳香环表面吸附的平衡几何构型。表2所示为气体分子在煤表面达到吸附平衡时的相关构型参数(其中,M代表煤分子,R代表原子之间的垂直距离)。由表1与表2的对比可以看出,O2和NH3在煤分子表面发生吸附前后,其键长、键角以及振动频率都未发生较大幅度的变化,符合物理吸附的特征。从吸附能大小比较可以看出,煤分子对O2的吸附性要小于对NH3的吸附性,其主要原因为煤分子与NH3和O2分子之间的作用为弱相互作用,且这种弱相互作用包括静电作用及范德华作用。对于上述两种弱相互作用,虽然他们的强度处于同一数量级,但前者的强度要大于后者。NH3为极性分子,O2为非极性分子,煤分子与NH3分子之间的相互作用主要为静电相互作用,然而煤分子与O2之间的相互作用除了一部分静电作用外,还存在大部分的相对较弱的范德华作用,因此煤分子对NH3的吸附能要大于对O2的吸附能。 表2 O2和NH3在煤表面不同位置吸附平衡几何构型参数对比Table 2 Comparison of geometric configuration parameters of O2 and NH3 adsorption equilibrium at different positions on coal surface 图4 O2和NH3在煤分子表面的能量最优吸附构型Fig.4 Energy optimal adsorption configuration of O2 and NH3 on coal molecular surfacea—O2 on aromatic ring surface;b—O2 on —NH2 side chain;c—NH3 on aromatic ring surface;d—NH3 on —NH2 side chain 本实验计算出的煤分子对O2和NH3的物理吸附能分别为-3.69 kJ/mol和-27.22 kJ/mol,这与文献[46]通过相关实验以及计算得出的煤与气体分子之间物理吸附的相互作用能绝对值范围为0 kJ/mol~40 kJ/mol的结论相符。通常化学吸附能远高于物理吸附能,文献[47]通过采用“多点计算,整体平均”方法计算得到O2在5种煤分子表面的化学吸附能达到-800 kJ/mol,这比本实验计算的物理吸附能要高出一个数量级,充分说明了本次计算结果为物理吸附。根据文献[48]的相关研究可知,煤表面与矿井采空区各种气体发生吸附的亲和顺序由大到小依次为:O2,N2,CH4,文献[49]通过研究算得CH4和N2在煤分子表面的吸附能为分别-2.78 kJ/mol和-1.08 kJ/mol,低于本实验计算的O2的吸附能,本次计算结果完全符合以上亲和性顺序规律。上述研究结果从不同侧面验证了本实验计算的正确性。 对于O2分子,其吸附在—NH2侧链时吸附体系能量最低,吸附构型最为稳定。对于NH3分子,其吸附在靠近氨基侧链的苯环位置时,吸附体系能量最低,吸附构型最为稳定。对于O2和NH3出现上述所示的最优吸附构型,其主要原因可以利用煤分子表面的静电势分布予以解释。对于O2分子,O原子电负性较大,呈现负电性,倾向于吸附在煤分子表面正静电势较大的区域,结合图2和图3,正静电势较大的区域为—NH2侧链上两个H原子周围区域,因此图4b为O2在煤分子表面的能量最优吸附构型。对于NH3,N原子呈负电性、H原子呈正电性,当NH3在煤分子表面吸附时,较多的带正电性的H原子倾向于吸附在煤分子表面中负静电势较大的区域,而N原子倾向于吸附在正静电势较大的区域,结合图2和图3,NH3分子会吸附在靠近—NH2侧链的芳香环表面,且两个H原子指向苯环的负静电势区域,N原子指向—NH2正静电势区域,该构型既满足了电子受体-电子供体的作用机制,又使得吸附质与吸附剂分子之间更易形成N—H…N和N—H…Np的氢键,因此如图4c所示的吸附构型最为稳定。 2.3.1 NH3在煤-O2混合体系的吸附 图5 NH3在煤-O2混合体系的吸附Fig.5 Schematic diagram of NH3 adsorption in coal -O2 mixed systema—Adsorption position;b—Adsorption distance and configuration diagram 表3 NH3在煤-O2体系能量最优吸附构型参数Table 3 Energy optimal adsorption configuration parameters of NH3 in coal-O2 system 2.3.2 O2在煤-NH3混合体系的吸附 图6 O2在煤-NH3混合体系吸附Fig.6 Schematic diagram of O2 adsorption in coal -NH3 mixed system 由表4可知,O2在煤-NH3混合体系的吸附能ΔE′值要远低于其单独吸附在煤分子表面时的吸附能ΔE′值,表明NH3的存在会极大的促进O2在煤分子表面的吸附,进而促进煤自燃氧化的进程。O2在煤-NH3混合体系的最佳吸附位置位于-NH2侧链,且同样遵循吸附能越大,吸附距离越短的特征。 表4 O2在煤-NH3表面能量最优吸附构型参数Table 4 Energy optimal adsorption configuration parameters of O2 on coal-NH3 surface 2.4.1 多个氧气分子在煤表面的吸附 表5 不同个数O2分子在煤分子表面吸附达到几何平衡构型时的键长和吸附能Table 5 Bond length and adsorption energy of different numbers of O2 molecule on coal surface when they reach geometric equilibrium configuration 图7 煤分子与不同个数的O2分子的吸附平衡几何构型Fig.7 Adsorption equilibrium geometric configuration diagram of coal molecules and different numbers of O2 moleculesa—1 O2 molecule;b—2 O2 molecules;c—3 O2 molecules;d—4 O2 molecules 2.4.2 煤与NH3和多个O2分子的混合吸附构型 煤表面与NH3和O2之间会发生混合吸附,混合吸附达到平衡时的几何优化构型如图8所示。将上述2.4.1已经模拟计算出的煤分子与不同个数O2分子吸附的能量最佳构型作为已吸附体系,通过加入NH3分子来研究其对已吸附体系的影响。 图8 煤分子与NH3分别与不同个数的O2分子的吸附平衡几何构型Fig.8 Adsorption equilibrium geometric configuration diagram of coal molecules and NH3 with different numbers of O2 molecules respectivelya—1 O2 molecule;b—2 O2 molecules;c—3 O2 molecules;d—4 O2 molecules 表6 NH3与O2分子在煤表面混合吸附达到最优吸附构型时的键长和吸附能Table 6 Bond length and adsorption energy of mixed adsorption of NH3 and O2 on coal surface to reach optimal adsorption configuration 1) 煤分子对NH3分子的吸附性强于对O2分子的吸附性。当单个气体分子在煤分子表面吸附时,NH3分子倾向于吸附在靠近—NH2侧链的苯环位置,而O2分子倾向于吸附在—NH2侧链,且煤表面吸附多个O2分子时,O2的吸附位置同样集中在—NH2侧链,表明煤的氧化自燃过程中侧链首先被氧化。 2) NH3在煤-O2混合体系的吸附能大于其单独在煤分子表面的吸附能,O2在煤-NH3混合体系的吸附能远大于其单独在煤分子表面的吸附能,表明NH3和O2分子在煤表面的吸附存在相互促进的作用,而NH3对煤分子吸附O2的促进作用要强于O2对煤分子吸附NH3的促进作用,这将加快煤的自燃氧化进程。 3) 对于煤分子表面吸附多个O2分子的情况,当O2与NH3的分子数比值为1~3时,该条件下NH3对煤吸附O2的促进作用随着O2分子数目的增加而显著增强,进而显著促进煤自燃现象的发生;当O2与NH3的分子数比值大于3时,NH3对煤吸附O2的促进作用有所减弱,但仍然为促进作用。1 煤结构模型的建立以及计算方法的选择
1.1 煤表面分子片段模型的选择

1.2 计算方法

2 结果与讨论
2.1 气体参数与煤分子表面的静电势分布



2.2 单个气体分子在煤表面的吸附



2.3 煤和NH3及O2混合体系吸附规律研究



2.4 煤与NH3,多个O2混合吸附的研究




3 结 论
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
天津理工大学学报(2021年2期)2021-06-03
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
少儿美术(2019年8期)2019-12-14
中国化妆品(2018年6期)2018-07-09
中学科技(2015年8期)2015-08-08
