
基于高维随机矩阵的癌症基因网络识别方法
2021-11-17 03:56任喜梅钟春晓王锦丽
计算机仿真 2021年3期
李 蓉,任喜梅,钟春晓,王锦丽
(华东交通大学理工学院,江西 南昌 330100)
1 引言
癌症由多阶段的多基因共同参与而生成、发展,并与基因变化有着紧密联系,癌症的发生、促进、发展以及转移,大部分都跟原癌基因[1]活化、抑癌基因[2]失活等基因突变密切相关。具有挖掘隐含生物学信息功能的微阵列数据,可以依据基因间的性能模块中已知基因预测未知基因,但因其维数较高,存在较少数量与癌症相关的基因。高维随机矩阵理论(Random Matrix Theory,RMT)通过对比随机的高维、多维序列属性特征,发现实际数据与随机因素之间的偏离程度,提取数据内的总体相关行为特征。
为此,本文将高维随机矩阵与癌症基因融合,提出一种癌症基因网络识别方法,将系数矩阵右边增加一列,扩增随机矩阵,提升随机矩阵特征拟合度;通过规范化、中心化以及标准化随机矩阵,提升矩阵适用性能;以互信息作为度量标准,依据各随机矩阵奇异值矢量与初始特征奇异值矢量的差值,优化特征选择;利用癌症基因表达矩阵特征根的最近邻间隔分布与高斯正态分布、泊松分布的标准误差比值计算,增加有效信息的保留数量,增强噪声滤除性能。
2 高维随机矩阵下癌症微阵列数据预处理
2.1 高维随机矩阵构建
将维度较高的随机变量作为矩阵组成元素,即可构成高维随机矩阵,依据随机矩阵理论含义,设定某一高维随机矩阵为M,表达式如下所示
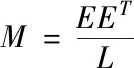
(1)
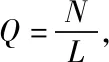
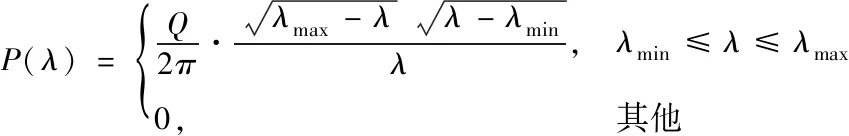
(2)
式中,随机矩阵M的极大、极小特征值分别用λmax和λmin来表示,相应表达式如下所示
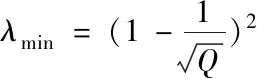
(3)

(4)
综上所述,利用随机矩阵M与相关矩阵C具有的属性特征,划分矩阵C为符合随机矩阵M部分与差异部分,即随机噪声U与真实信息V,通过优化相关矩阵C,即可去除其中所含噪声。
2.2 癌症微阵列数据特征选择优化
基因表达网络[3]受实验条件影响,一般会存在一些随机因素:当实验时间与样本条件发生变化时,基因表达水平也将随之改变;若实验样本有限,则有可能产生测量噪音。而此类随机因素生成的虚假信息,会对真实信息造成干扰,影响识别结果的可靠性与准确性,因此,应在初始阶段去除可能产生的随机因素。
已知矩阵D是一个初始的数据矩阵,由特征集合F={f1,f2,…,ft}与类集合S={s1,s2,…,sk}架构而成,其中,t、k分别表示特征个数与类别数量,高维随机矩阵M的构建公式如下所示
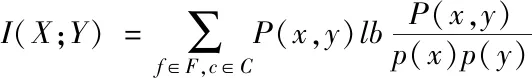
(5)
下列矩阵即为所得矩阵M的表达式
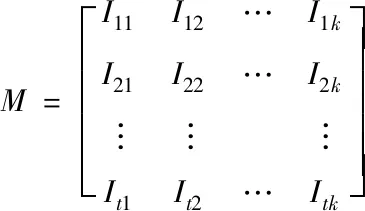
(6)
若上式中的k值较小,则无法理想地满足随机矩阵特征,所以,应在系数矩阵右边增加一列,扩增[4]随机矩阵M,复制m次后,得到下列表达式
M=[M,m(M)]
(7)
式中的m可通过下列计算公式完成求解,令初始的行列比值不发生改变
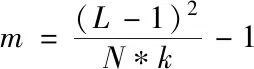
(8)
为确保该随机矩阵M并非一种特例,可以代表大多数的普遍情况,采用下列式(9)与(10),规范化、中心化以及标准化随机矩阵M,得到不失一般性的随机矩阵Md
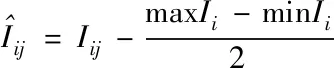
(9)
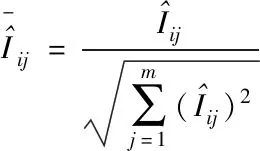
(10)
利用得到的随机矩阵Md,推导出t×t的特征相关矩阵C表达式,如下所示

(11)
再通过下列奇异值分解[5]式,完成相关矩阵C的奇异值分解
C=UΛV
(12)
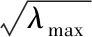
Cnew=UΛnewV
(13)
任意两特征与初始类别的关联程度,用Unew中含有的各项元素Kij来描述,各特征与新类别的关联程度,用Vnew中含有的各项元素Eij来描述。通过对去噪的相关矩阵进行特征选择,可去除冗余以及与类别不存在关联性的特征。因为去噪后留存了j-1个奇异值,且选取特征数量与奇异值剩余个数相同,所以,特征选择个数是j-1,各特征的重要程度计算公式如下所示
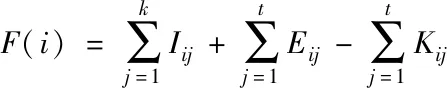
(14)
式中,第i个特征的重要程度为F(i),集合是F={f1,f2,…,fi},按照从大到小的顺序降序排列F(i)后,得到重要程度最高的前j-1个特征,完成特征选取。
采用随机矩阵进一步优化选择的n个特征,各特征均是一个随机变量,利用随机特征矩阵与初始特征矩阵奇异值矢量的相关系数,描述特征与随机变量的关联度,相关系数越大,关联度越高,所以,应留存较小的相关系数特征。
已知特征集合F={f1,f2,…,fn},类别数量为k个,计算初始特征矩阵M的奇异值过程中,以互信息[6]作为度量标准,采用式(5)和(6)架构互信息矩阵D,再依据式(9)~(12),逐步实施规范化、标准化、相关矩阵运算以及奇异值分解等操作,最终得到矩阵M的奇异值矢量e,该矢量的组成部分为n个奇异值,其中所含元素表示为εk;在求取随机特征矩阵奇异值矢量阶段,将随机变量用各个特征表示,构建数量为n的随机矩阵(M1,M2,…,Mn)。按照初始特征矩阵奇异值矢量计算流程,解得各随机矩阵Mi(1≤i≤n)奇异值矢量ei,其中所含各元素用εik表示。关于各随机矩阵奇异值矢量ei与初始特征奇异值矢量e的差di,可利用下列计算公式解得,通过取整差值di,并保留di≠0情况下的fi,实现特征选择优化
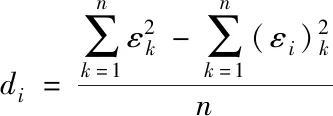
(15)
3 癌症基因网络识别
3.1 降噪点确定
利用随机矩阵理论与下列皮尔森相关系数[7]公式,转换癌症基因微阵列数据为相关基因矩阵,使矩阵中含有全部基因之间的关联程度:
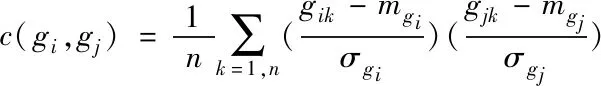
(16)
假设高维随机矩阵理论的降噪参数是q,取值范围为(0,1),将参数q值慢慢增大,去除较小的相关系数,求解对应于各降噪参数q的特征根NNSD(Nearest-neighbour Spacing Distribution,最近邻间隔分布)。在不断增大参数q值、去除相关矩阵的较小相关系数过程中,相关矩阵特征根最近邻间隔分布形式由高斯正态分布过渡至泊松分布[8]。
通过标准误差方法,可以准确、科学地确定癌症基因特征根分布形式过渡至泊松体系的转变点对应q值,所以,设定癌症基因表达矩阵特征根的最近邻间隔分布与高斯正态分布的标准误差为SDGOE(q),与泊松分布的标准误差为SDpoisson(q),利用下列两项标准误差界定公式,计算分布形式过渡的临界点与降噪参数
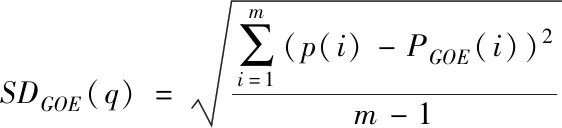
(17)
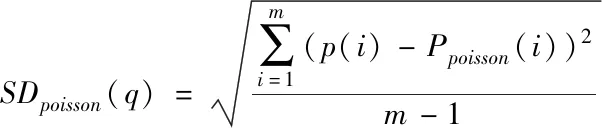
(18)
式中,第i点对应特征根的最近邻间隔分布是p(i),该点对应高斯正态分布与泊松分布特征根的最近邻间隔分布分别用PGOE(i)、Ppoisson(i)表示。
如果两个标准误差值相同,特征根的最近邻间隔分布体系元素相互效用较强,相关程度较大,相似性较高,多数为真实相关信息,极少数为随机信息;如果标准误差比值较大,分布体系则更趋近于泊松分布,偏离高斯正态分布,在留存有效信息的同时,充分滤除噪声。因此,降噪点即为最大标准误差比值的对应点。将降噪参数q从0逐渐增大至1,去除癌症相关矩阵含有的随机噪声,才能得到真实的癌症基因网络。
3.2 癌症基因网络架构
依据癌症基因初始数据与不同实验条件的所有基因表达水平,采用cluster tree view软件构建描述基因相互效用的层次树形图,该图在聚合有相关性基因的同时,展现出层次结构的连接形式。基于明确的降噪点,选取出与其它基因相关系数不小于降噪参数q的基因,构建新的癌症基因表达矩阵,并聚类分析经过噪声滤除的癌症基因。
假设新建癌症基因表达矩阵的留存基因数量为361,且新矩阵的基因内涵盖其中的所有信息,则该癌症基因网络树形图如图1所示。
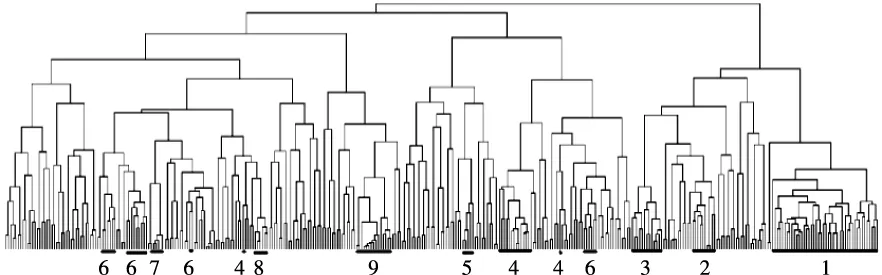
图1 癌症基因网络树形图
树形图1中,基因之间的相关性用连线表示,相关程度的强弱用树枝长度表示;连线与相关程度成反比。
4 实例分析
4.1 实验数据采集
实验环节以肝癌[9]为例,从http:∥ genome-www.stanford.edu hcc supplement.shtml.斯坦福微阵列数据库中,挑选肝癌基因微阵列初始数据,得到的研究数据为基于82个HCC(Hepatocellular Carcinoma,肝细胞性肝癌)样本1648个肝癌基因的微阵列数据,样本基因信息如表1所示。

表1 肝癌基因数据统计表
4.2 降噪点获取
从0到1逐渐增大高维随机矩阵理论的降噪参数q,图2所示为参数q取不同数值时,肝癌基因特征根的最近邻间隔分布情况,图中泊松分布用虚线表示,高斯正态分布用实线表示,最近邻间隔分布用点线表示。
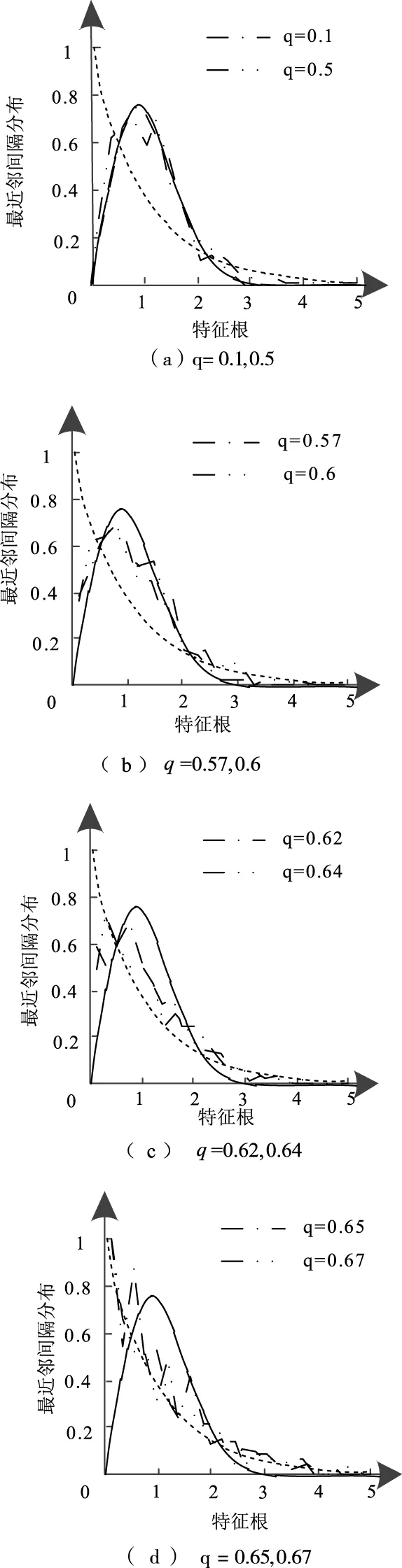
图2 不同参数值的最近邻间隔分布变化
通过图2的曲线走势可以看出,参数q在数值到达0.64后(见图2(c)),基因特征根最近邻间隔分布变化趋势开始趋于泊松分布,经过标准误差值运算,发现参数q在取值是0.67时(见图2(d)),标准误差比值为极大值,因此,1648×82肝癌微阵列数据的降噪点参数取值为0.67。
4.3 癌症基因网络识别结果
去噪后保留820个肝癌基因,并得到820×82的微阵列数据,利用Cluster 3.0软件层次聚类肝癌基因,采用斯皮尔曼相关系数度量基因之间的相似性,通过cluster tree view软件呈现的肝癌基因树形图,如图3所示。
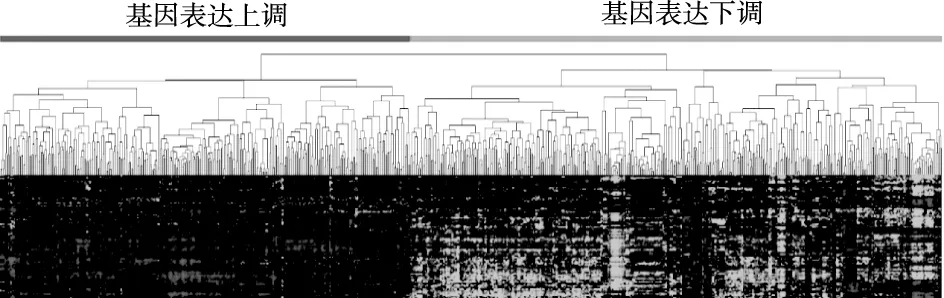
图3 基因树形图
图3的基因表达谱中,基因表达量上调用红色表示,下调则用绿色表示。当基因被划分为一类时,相同肝癌样本的表达量上、下调一致。经分析得到的团簇分别是增殖簇、B淋巴细胞簇、细胞周期调控簇、基质细胞簇以及脂类酒精代谢簇,如图4所示。
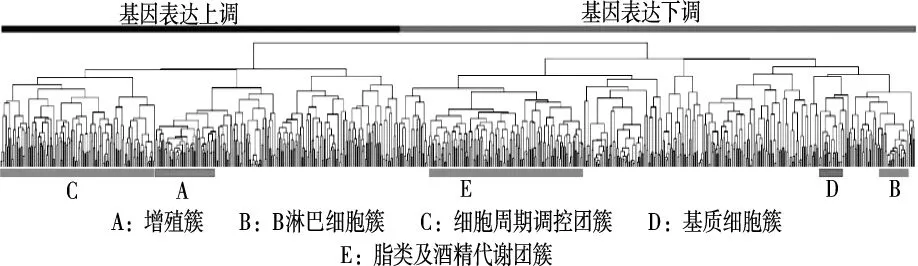
图4 肝癌基因团簇分类示意图
以关联程度较强的B淋巴细胞簇为例,分析肝癌基因团簇,图5所示为B淋巴细胞簇的基因树形图。从B淋巴细胞团簇中分别识别出B淋巴细胞的增长因子WNT4与编码mRNA前剪接调控因子SLU7两个基因。在团簇内层,两基因紧密连通,该基因与B淋巴细胞免疫过程相关,说明淋巴细胞正浸润肝组织。
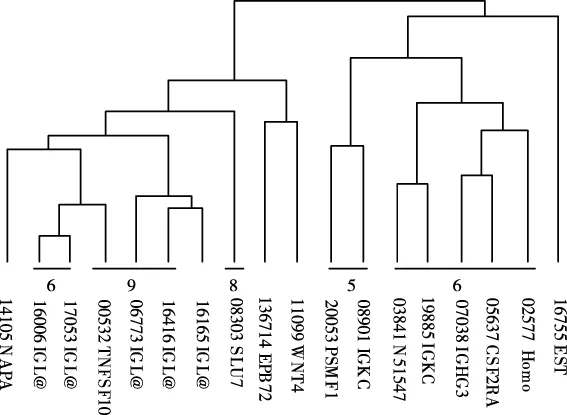
图5 B淋巴细胞簇基因及其层次树图
为了验证本文方法识别的有效性,采用基因网络模块划分方法[10],得到图6所示的肝癌基因处理结果。

图6 基因网络模块划分方法下肝癌基因模块图
经过对比图5和图6可以看出,基因网络模块划分方法识别出的肝癌基因中,只有1个基因在本文方法构建的树形图分支上与对应的主分支有所偏离,其它相同模块的基因均与本文树形图所属分支一致,说明本文方法能够识别出基因的真实模块,且相似度较高。
5 结论
癌症作为一种复杂性疾病,对人类健康存在严重威胁,只有及时查出癌症发生的相关基因,发现互相关联,才能防止癌症恶化,因此,本文针对癌症基因的微阵列数据,以高维随机矩阵为数据预处理策略,提出一种癌症基因网络识别方法,并制定出今后的研究探索方向:在过渡至泊松分布的过程中,最近邻间隔分布体系仍有可能存在噪声干扰,需设计出一个更加优化的降噪点判定方法,使噪声能够去除完全;为便于基因的后续调控,需量化模块之间的连接大小与调控关系,并进一步研究基因之间相互调控的形式与力度,以及基因间相互影响程度。该方法对癌症的生成检测、恶化控制与治疗,有着重要的现实意义。
猜你喜欢
导航定位学报(2022年4期)2022-08-15
计算技术与自动化(2022年1期)2022-04-15
北京航空航天大学学报(2021年9期)2021-11-02
中学生数理化·高一版(2021年11期)2021-09-05
科学导报(2020年85期)2020-01-13
世界知识画报·艺术视界(2017年7期)2017-07-27
今日中国(2017年3期)2017-03-31
新东方英语(2016年4期)2016-04-06
新高考·高一物理(2016年1期)2016-03-05
读写算·小学低年级(2014年4期)2014-07-24
