
基于表示学习和深度森林的长链非编码RNA编码短肽预测模型
2022-01-05 02:32纪腾其赵思远胡鹤还
计算机应用 2021年12期
纪腾其,孟 军,赵思远,胡鹤还
(大连理工大学计算机科学与技术学院,辽宁大连 116024)
(∗通信作者电子邮箱mengjun@dlut.edu.cn)
0 引言
长链非编码RNA(long non-coding RNA,lncRNA)作为非编码RNA(non-coding RNA,ncRNA),能够调节动物[1]与植物[2]的生长、发育、疾病等一系列生命活动。然而,最近有研究表明一些lncRNA 中含有长度不超过300 bp 的小开放阅读框(small Open Reading Frames,sORFs),具有编码蛋白的能力[3],能翻译一类长度不超过100个氨基酸的短肽[4]。这类由lncRNA 上的sORFs 区域编码的短肽(sORFs-encoded short peptides,SEPs)在生物的各种生命活动中发挥重要作用[5-6]。
SEPs 的发现打破了人们以是否编码区分mRNA 与ncRNA的标准,显著地扩大了蛋白质组的范围和多样性,随着越来越多的短肽及其功能的发现,人们也开始将视线集中到对短肽的分析识别上[7]。
基于机器学习的计算预测方法在各种领域中都有出彩的表现,在生物信息领域中的lncRNA 识别和功能预测[8]、lncRNA-蛋白质相互作用[9]和lncRNA-miRNA 相互作用[10]等多个研究方向都取得了不错的成效。许多研究工作利用机器学习算法构建预测模型,通过将序列特征、结构特征或者其他方式的编码作为输入数据,构建分类器模型。
目前,植物lncRNA 编码短肽的识别与研究尚属新兴的研究领域,对于植物lncRNA 中的sORFs 以及对应的SEPs 的结构信息、理化特性的分析还不充分,而拟南芥作为经典的模式植物,经常被用于生物研究中[11],具有代表性,因此,以拟南芥的编码序列(Coding Sequences,CDS)和非编码序列(Non-Coding Sequences,NCDS)为切入点,对sORFs 序列的特征进行系统性分析,考虑到sORFs特征不鲜明、数据样本不足的性质,提出一种自编码器(AutoEncoder,AE)与深度森林(Deep Forest,DF)结合的方法,实现lncRNA编码短肽的识别预测。
本文的主要工作如下:
1)使用多个生物信息学软件获取拟南芥中的sORFs,对结果取交集以提高结果可信度;
2)提取CDS 与NCDS 候选sORFs 序列在多种特征编码方式下的特征表达结果,对特征进行融合,并通过机器学习算法分析二者的差异;
3)结合AE非监督表示学习思想,提出一种DF预测模型,实现sORFs编码短肽的识别预测。
1 相关工作
对植物lncRNA 编码短肽的识别本质上是识别植物lncRNA 中sORFs 是否具有编码能力,目前已有一些生物信息学工具可以用于挖掘sORFs[12-13]。如ORF finder[12]通过执行六个可读框的翻译,分析所有可能的ORFs 区域;sORF finder[13]基于六聚体组成偏差来实现对sORFs的识别。
目前对于SEPs 的研究大多通过生信工具获取到候选sORFs 作为数据支撑,通过生物实验、计算方法等进行分析与研究[5,14]。如Fesenko 等[5]通过质谱数据注释苔藓lncRNA 中的候选sORFs,使用BLAST 工具搜索具有同源序列的保守sORFs,并通过生物实验得到高可信度的SEPs,验证生物学功能。Zhu 等[14]为避免繁杂的生物实验,根据sORFs 序列差异性,使用逻辑回归模型完成SEPs 的识别,该机器学习方法代价低耗时少,但由于数据及特征表达不全面,无法有效预测较长的SEPs。
AE 是一种通过无监督学习学到主要特征并依据主要特征重构输入数据的人工神经网络,基于AE的表示学习能够有效地获取输入数据的高效表示[15],在各个领域得到了广泛应用[16],也能够很好地解决生物信息领域的问题[17]。近年来,深度神经网络(Deep Neural Network,DNN)在自然语言处理、视觉识别和生物信息等领域取得巨大成功[18],但伴随着训练数据量和超参数调优技能的高要求,而DF 作为DNN 的有效替代方法[19],只需较少的超参数,在解决生物信息问题上取得了较好的成果[20]。
本文提出了一种基于AE 表示学习和DF 的预测模型,不仅避免了无法充分提取特征的弊端,也不受到当前SEPs研究中有效数据量不足的影响,实现了对SEPs的有效识别预测。
2 数据预处理
本章将介绍数据集构建和特征编码方式,并对特征进行系统性分析。
2.1 数据集构建
由于经过生物实验验证的SEPs 数据稀少,无法满足机器学习的需求,因此通常采取特定方式合理构建数据集。从公共数据库Phytozome[21](https://phytozome.jgi.doe.gov/pz/portal.html)下载拟南芥CDS 数据和NCDS 数据,分别作为正集和负集的数据源。
对CDS使用sORF finder(http://hanadb01.bio.kyutech.ac.jp/sORFfinder/)和ORF finder(https://www.ncbi.nlm.nih.gov/orffinder/)获取sORFs,并对两种工具的结果取交集,之后通过CD-HIT[22]工具,去除相似度高于80%的序列[23],得到候选的正集sORFs 数据(图1(a))。由于sORF finder 寻找的是有编码能力的sORFs,因此对NCDS 只使用ORF finder 工具获取其中的sORFs,得到的结果通过相同的去冗余处理(图1(b))。考虑到目前发现能够编码的sORFs序列通常以碱基组合ATG 作为起始密码子,因此从去冗余后的结果中筛选出起始密码子为ATG 的sORFs 作为候选的负集sORF 数据。获取候选sORF的流程如图1所示。
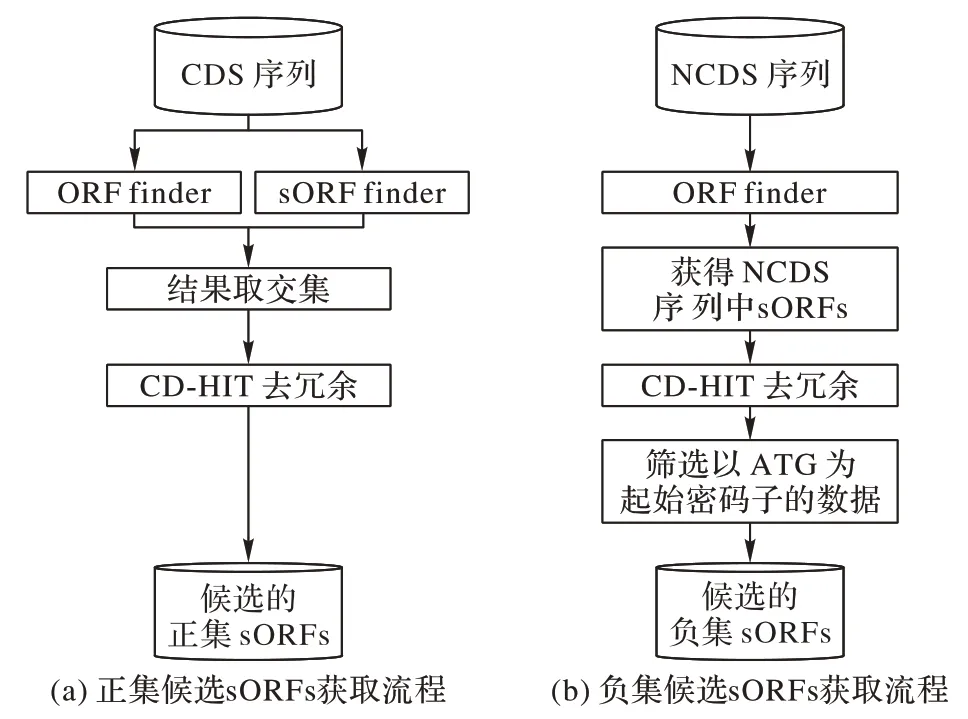
图1 候选sORFs的获取流程Fig.1 Process of obtaining candidate sORFs
经过以上处理得到CDS 和NCDS 的sORFs 数据分布差异较大,为了得到相似的正负集数据,对两部分数据进行了同分布取样,最终得到sORFs正负集数据各4 800条。
2.2 特征编码
考虑到SEPs独有特征不鲜明,本文采用传统的RNA特征提取方法提取了sORFs序列的相关特征。
序列的原始表达具有生物学意义,基于原始表达可以提取序列长度L、(G+C)碱基含量GC_content与GC 碱基的比例GC_ratio三个特征,融合后特征记为Feature1:

其中,G_num和C_num分别为序列中碱基G和碱基C的数目。
密码子是遗传物质编码的信息规则,CDS 与NCDS 的密码子保守性表现有所不同,因此提取k-mer特征作为密码子频率的近似表达,k代表序列中相邻的碱基数,k个相邻碱基有4k种组合。由于sORFs序列较短,因此取k=1,2,3。提取方法为沿sORFs 序列使用长度为k、步长为1 的滑动窗口进行滑动匹配,为避免3-mer 与1-mer 计算时因使用出现次数而导致较大差异,为每个k-mer特征分配权重,则有:
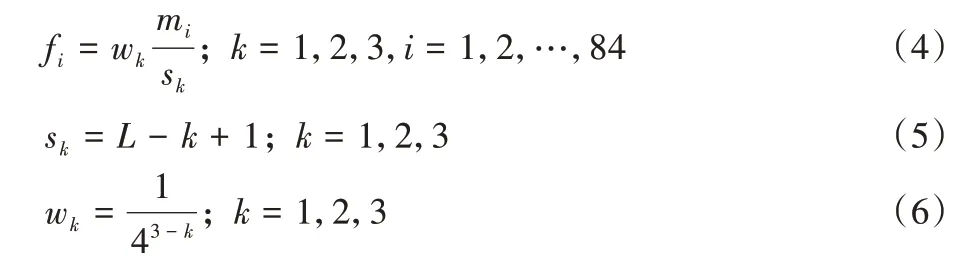
其中:wk为权重;sk为滑动次数;mi为每个k-mer 出现次数;fi为每个k-mer 经过归一化的频率,最终得到84 个k-mer 特征,记为Feature2。

k-mer特征仅考虑连续碱基的性质,然而不连续碱基也可能存在差异,采取短序列模体(Short Sequence Motifs,SSM)特征作为补充,相隔1 个碱基的碱基组成记为N*M,相隔2 个碱基的碱基组成记为N**M,N 与M 可以为A、T、C、G 中的任意一个碱基,每组短序列模体都有42维特征表达,提取N*M、N**N与N***M三组短序列模体特征:

其中:vk为滑动次数;ni为每个短序列模体出现次数;SSMi即为每个短序列模体的出现频率,最终得到48 个SSM 特征,记为Feature3。
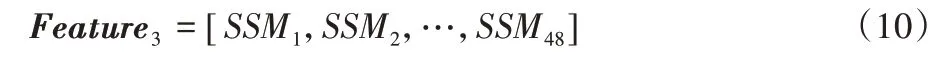
信噪比的大小能够表示CDS 和NCDS 中碱基使用的偏向性[24],通过碱基在密码子三个相位的分布计算sORFs 三分之一处的功率谱计算信噪比R:

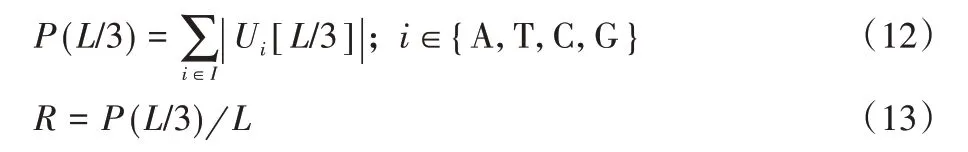
其中:xi、yi、zi为碱基i的一种在三个相位出现的频数的一行三列的数组;P(L/3)表示L/3处的功率谱。
最终,将上述特征组成136 维的特征集,将其作为AE 的输入向量Feature:

2.3 特征可视化分析
为验证特征编码的有效性,对其中维数较多且占比较大的k-mer 和SSM 特征进行了可视化分析,结果如图2 所示,可视化分析方法分别选取了主成分分析(Principal Component Analysis,PCA)、核主成分分析(Kernel Principal Component Analysis,KPCA)、t-分布邻域嵌入(t-distributed Stochastic Neighbor Embedding,t-SNE)和奇异值分解(Singular Value Decomposition,SVD)。从图2 中可以看出,降维之后的k-mer特征与SSM 特征较为明显地分布于不同的区域,说明CDS 与NCDS中sORFs存在一定的差异,也验证了上述特征编码方式的有效性。
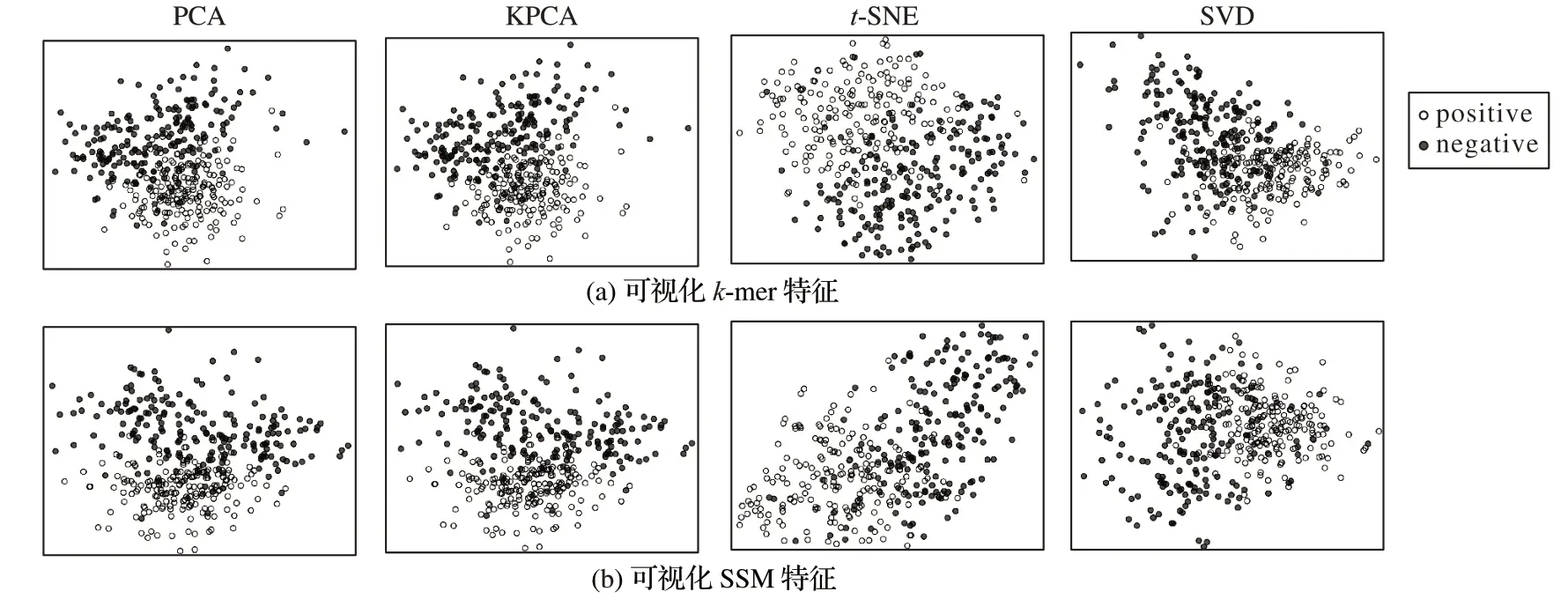
图2 不同特征编码方式的特征可视化结果Fig.2 Feature visualization results for different feature encoding methods
3 基于自编码器的深度森林模型
本文模型由特征编码、AE和DF三个阶段组成。
3.1 自编码器表示学习阶段
AE是典型的无监督机器学习方法,使用大量函数和神经网络结构产生高维输入的低维表示,由编码器与解码器两部分组成。AE的流程如图3所示。

图3 AE流程Fig.3 Flowchart of AutoEncoder
AE 接收输入的数据向量x后,经过编码器的多个隐藏层对其进行线性变换,在激活函数的作用下得到编码向量y,之后该向量经过解码器的多个隐藏层的变换后,得到重构之后的输出向量z,通过对比x与z,求出预测误差并反向传递,反复迭代得到最优权重:

其中:fθ是参数为θ={W,b}的编码层线性函数;gθ′是参数为θ′={W′,b′}的解码层线性函数;W是一个d′×d的权重矩阵,W′是W的转置,b和b′则是偏倚向量;s是激活函数。
由于ReLU 函数具有便于稀疏化和能够有效减小梯度似然值的优势,因此在编码与解码过程中都选择ReLU 函数作为激活函数。
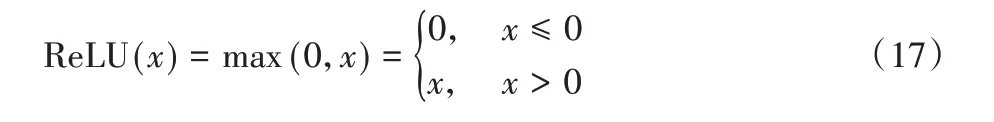
编码器设置了3 个隐藏层,每层的神经元数量逐层减少;解码器设置了3 个隐藏层,每层的神经元数量逐层增加;选取AE 的中间隐藏层的输出作为最终的表示学习结果。考虑到不同维数的表示学习结果对于分类器性能的影响也不同,因此,通过改变中间隐藏层神经元数量来寻找较优的表示学习结果,不同维数的表示学习结果训练得到的分类器以准确率为评价指标,结果如图4 所示。从图中可以看出,在表示学习结果维数为75时,性能相对更优。

图4 不同维数特征的分类准确率Fig.4 Classification accuracy of features with different dimension
3.2 深度森林训练阶段
DF 是随机森林(Random Forest,RF)[25]的扩展算法,借鉴神经网络的特性,具有高效率、低数据量和低超参数要求等优势,由多粒度扫描和级联森林组成[19]。
在多粒度扫描阶段,DF 采用不同大小的滑动窗口对输入数据进行采样,以此作为级联森林的输入。经实验最终选取长度分别为2 和4 的滑动窗口进行采样,挖掘更加全面的信息。
在级联森林阶段,第一层级联以多粒度扫描中的第一个滑动窗口扫描得到的结果作为输入,通过多个RF 进行训练,产生增强特征,并将增强特征与经过转换的多粒度扫描中第二个滑动窗口得到的特征向量拼接并传递给下一层级联,下一层级联以拼接向量作为输入,重复上述过程。在每一层级联产生新的增强向量后,都在验证集上进行验证,如果验证得到准确率有所提升,则将增强向量继续传递给下一层的级联,产生新的拼接向量;如果没有提升,则终止训练。级联森林阶段每一层级联由4个RF组成,每个RF包含1 000棵决策树。
3.3 模型实现
本文模型由特征编码、AE 和DF 三部分组成。首先通过提取sORFs 相关特征完成编码;紧接着将结果输入到AE 中,经过ReLU 函数激活,完成表示学习过程;最后将表示学习得到的特征向量输入到DF 中,经过自适应的层数完成训练,获得每一维增强特征对应的分类概率,通过argmax 函数得到最终的分类结果,完成预测。模型的具体结构如图5所示。

图5 模型整体结构Fig.5 Overall structure of model
4 实验与结果
用拟南芥数据集对比不同方法的性能,验证模型的预测能力,并在大豆和玉米数据上进行测试,验证模型的泛化能力。
4.1 评价指标
实验以准确率ACC(Accuracy)、精确率P(Precision)、召回率R(Recall)和F1 值F1(F1_score)作为评价指标。四种评价指标的计算公式如下:

其中TP、FP、TN、FN的含义如表1所示。

表1 分类结果含义Tab.1 Meaning of classification results
4.2 对比传统机器学习模型的分类结果
在拟南芥数据集上将提出方法与朴素贝叶斯(Naive Bayes,NB)[26]、决策树(Decision Tree,DT)[27]、随机森林(RF)、自编码器(AE)与三种模型结合的组合模型以及DF 进行比较。实验结果如表2 所示。可以看出,本文模型在准确率、精确率、召回率和F1 值四个指标都优于其他传统机器学习模型,说明本文模型在预测SEPs 上具有良好的分类性能。在准确率方面分别比AE+NB,AE+DT,AE+RF 模型高15.31、5.72、4.58 个百分点,说明DF 模型的性能优于其他模型。同时,本文模型的准确率也较使用单一DF作为分类模型提高了4.16 个百分点,验证了AE 表示学习的有效性,能够学习到的特征以更少的维数取得了更高的性能。此外,从最小显著性差异法分析结果可以看出,本文模型显著优于传统机器学习模型且准确率的标准差SD(Standard Deviation)仅为1.2%,表明模型的稳定性较好。

表2 本文模型与传统机器学习模型及其组合模型以及DF在拟南芥数据集上的结果比较Tab.2 Result comparison of the proposed model with traditional machine learning models,their combined models and DF on Arabidopsis thaliana dataset
4.3 对比深度学习模型的分类结果
除了与传统机器学习模型进行对比,还将本文模型在拟南芥数据集上与深度学习模型进行对比,如卷积神经网络(Convolutional Neural Network,CNN)[28]、递归神经网络(Recurrent Neural Network,RNN)[29]以及AE 与它们相结合的组合模型,实验结果如表3 所示。从表3 中可以看出,与深度学习模型相比,本文模型在准确率方面分别比CNN、AE+CNN、RNN 和AE+RNN 模型高1.66、1.04、2.29 和2.08 个百分点,说明本文模型具有良好的分类性能;同时,本文模型以DF作为训练模型,具有低数据量与低超参数调优技能要求的优势。
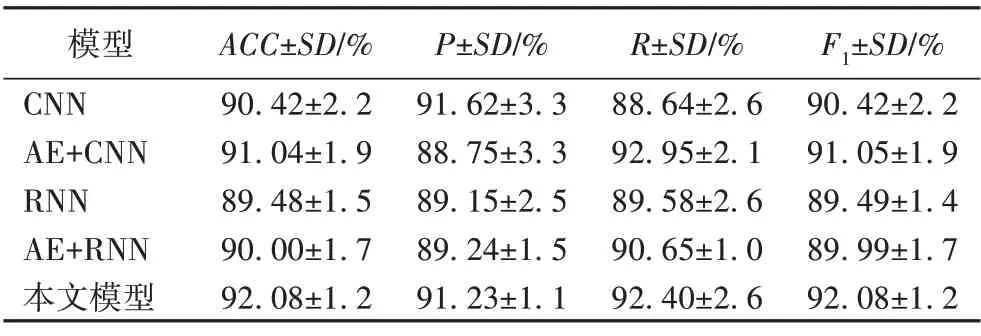
表3 本文模型与深度学习模型及其组合模型在拟南芥数据集上的结果比较Tab.3 Result comparison of the proposed model with deep learning models and their combined models on Arabidopsis thaliana dataset
4.4 不同物种上的分类结果
为了验证模型的泛化能力,在其他物种数据集上也对本文模型进行了测试[10]。从Phytozome 数据库下载大豆(Glycine max)和玉米(Zea mays)的CDS 与NCDS 数据,并进行与拟南芥数据集相同的预处理,并从处理完毕的数据集中随机选取与拟南芥数据集具有相似分布的大豆和玉米数据作为独立测试数据集。实验结果如表4 所示,可以看出,本文模型在玉米和大豆lncRNA 编码短肽预测方面各个指标都较好,说明该模型具有良好的泛化能力,能够适用于其他物种。

表4 本文模型在大豆和玉米数据集上的分类结果Tab.4 Classification results of the proposed model on Glycine max and Zea mays datasets
5 结语
通过多种特征编码处理序列,结合特征降维方法,对拟南芥CDS 与NCDS 中的sORF 进行可视化分析,证明特征编码的有效性。综合考虑sORFs 特征不鲜明且相关数据偏少的特点,提出一种基于自编码器(AE)和深度森林(DF)的SEPs 识别模型。实验结果表明,与多种模型相比,本文模型具有更优的性能;此外,在大豆与玉米数据集上进行独立测试,也取得了良好的效果,验证了该模型良好的泛化能力,能够适用于其他物种。未来将尝试使用更多的方法进行更深入的表示学习来进一步改善对lncRNA编码短肽的预测能力。
猜你喜欢
电工技术学报(2022年20期)2022-10-29
科技视界(2022年17期)2022-08-10
客联(2022年3期)2022-05-31
科学之谜(2021年2期)2021-04-25
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
飞碟探索(2015年9期)2015-11-05
红领巾·探索(2015年9期)2015-09-10
恋爱婚姻家庭·养生版(2011年8期)2011-05-14
