
UPLC-MS/MS法测定牛奶中氯霉素残留量的不确定度评定
2022-02-09 13:35李绍群严国荣周雨薇刘东方
现代盐化工 2022年6期
李绍群,严国荣,李 方,周雨薇,刘东方
(1.兴化市食品质量检验检测中心,江苏 兴化 225700;2.新沂市瓦窑中学,江苏 徐州 221438)
氯霉素(Chloramphenicol,CAP)的分子式为C11H12CL2N2O5,是氯霉素类抗生素的代表药物,主要通过抑制细菌蛋白质的合成起到抗菌的作用,抗菌谱比较广,包括G+和G-,对伤寒杆菌、百日咳杆菌等均具有较强作用[1]。过量使用CAP会产生严重的毒副作用,临床应用因此受到限制[2]。
测量不确定度(Measurement Uncertainty,MU)简称不确定度,表示与测量结果相关联的参数,表征被测量的量值分散程度[3]。一般情况下,不确定度越低说明检测水平越高,检测结果越可靠[4]。
样品经提取、净化、内标法定量,采用超高效液相色谱-串联质谱法(Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry,UPLC-MS/MS)处理,其准确性易受到样品处理、实验环境、曲线拟合和仪器设备稳定性的影响,测量数据难免产生误差。本研究根据相关标准并参考已发表的文献,对测定牛奶中CAP质量分数方法的不确定度进行评定。
1 试验
1.1 材料与试剂
甲醇中的CAP(坛墨,100 μg/mL,1.2 mL);乙腈D5-CAP(内标,坛墨,100 μg/mL,1.2 mL);乙腈、甲醇(均为色谱纯);氨水、乙酸乙酯(均为分析纯);实验用水为自制超纯水;亲水亲脂平衡(Hydrophile Lipophilic Balance,HLB)固相萃取柱(安谱,60.00 mg,3.0 mL);含CAP的牛奶质控样品。
1.2 仪器与设备
UPLC-MS/MS(TQS/I-Class,Waters);涡旋仪(DL-866,IKA);电子分析天平(FA3204B,万分之一);台式高速冷冻离心机(3K15,SIGMA);氮吹仪(TTL-DCI);超纯水系统(Milli-Q);移液器(100~1 000 μL、20~200 μL和2~20 μL,Eppendorf);不同规格量瓶。
1.3 试验方法
1.3.1 样品提取与净化
准确称取样品(2.00±0.02)g于50.0 mL离心管中,加入内标D5-CAP标准工作溶液50 μL,准确加入10.0 mL 95%氨化乙酸乙酯(乙酸乙酯与氨水的体积比为95∶5),振荡5 min,10 000 r/min离心3 min,量取上清液5.0 mL,40 ℃氮气吹干,残渣用0.5 mL甲醇复溶,振荡1 min,2.0 mL水稀释混匀,待净化。
取HLB固相萃取柱(60.00 mg,3.0 mL),依据说明书进行净化。量取0.5 mL 50%乙腈水溶液复溶,涡旋1 min,过滤待分析。
1.3.2 标准工作溶液的配制
100.0 μg/L D5-CAP标准工作溶液:精确量取100 μg/mL D5-CAP标准溶液1.0 mL,10倍梯度逐级稀释至目标质量浓度,有效期3个月。
50.0 μg/L CAP标准工作溶液:精确量取100 μg/mL CAP溶液1.0 mL,逐级稀释至目标质量浓度,有效期1个月。
1.3.3 系列基质标准溶液配制
精确量取适量CAP标准工作溶液和50 μL D5-CAP内标工作溶液,分别加入6份经提取、净化的空白样品中,40 ℃氮气吹干,精确量取适量50%乙腈水溶液复溶,配制成CAP质量浓度依次为0.1、0.2、0.5、1.0、2.0、5.0 μg/L,含D5-CAP内标质量浓度为5.0 μg/L的系列基质标准溶液,过滤待分析。
1.3.4 超高效液相色谱-串联质谱条件
色谱和质谱条件参考文献[5]进行优化。数据采集和处理软件为MassLynx 4.0。
1.4 数据采集及模型建立
以得到的目标物与内标峰面积比值y与相应的CAP质量浓度x绘制标准曲线,通过最小二乘法拟合得标准曲线y=ax+b,a为斜率,b为截距。
样品中CAP质量分数计算公式:

式中:X为样品中CAP的质量分数,μg/kg;ρ为样品溶液中CAP的质量浓度,μg/L;m为供试品称样质量,g;f为样品溶液稀释因子。
1.5 不确定度来源分析
利用UPLC-MS/MS法测定牛奶中CAP的质量分数,根据检测过程分析可知,测定不确定度的来源主要有样品的称量、提取与净化、实验环境、标准品纯度、系列标准溶液配制、标准曲线拟合、加标回收和重复精密度等因素。
2 不确定度结果与分析
2.1 样品称量引入的不确定度urel(m)
试样称量用电子天平感量为0.01 mg,由计量证书可知,天平的示值误差为±0.05 mg,称量不确定度服从均匀分布,其标准不确定度:

样品称量质量W为2 024.00 mg,则样品称量引入的相对标准不确定度:
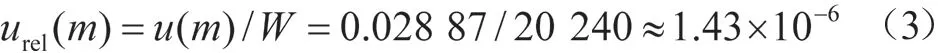
2.2 样品前处理过程引入的相对标准不确定度urel(S)
2.2.1 样品前处理过程中量器引入的不确定度urel(V1)
吸量管引入的不确定度由吸量管校准与温度效应引入的不确定度组成。前处理用吸量管均为A级,其量程V为10.0、5.0、2.0、1.0、0.5 mL,查阅JJG 196—2006《中华人民共和国计量检定规程 常用玻璃量器》[6]可知,A级吸量管在20 ℃下的容量允差如。按照三角形分布考虑,
10.0 mL量器引入的相对标准不确定度:
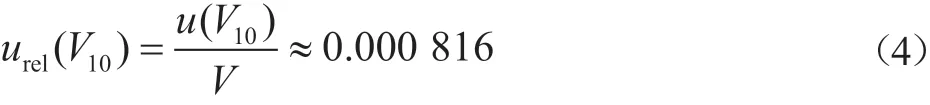
其他规格吸量管依上述过程进行计算。
2.2.2 样品前处理温度引入的不确定度urel(T1)
样品前处理过程中所用玻璃量器在20 ℃下校准,实验室温度一般在(20±5)℃,由于玻璃量器受温度影响较小,忽略不计。本实验前处理用试剂有乙腈、乙酸乙酯、甲醇和水,溶剂体积膨胀系数a分别为1.37×10-3/℃、1.39×10-3/℃、1.18×10-3/℃、2.08×10-4/℃,则50%乙腈水溶液的体积膨胀系数为7.89×10-4/℃。
在配制样品溶液时,实验室温度为22 ℃,温度变化按矩形分布时,以量取10.0 mL乙酸乙酯为例,温度效应引入的标准不确定度为u(T10):
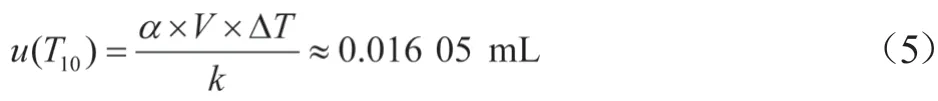
温度引起的相对标准不确定度为urel(T10):
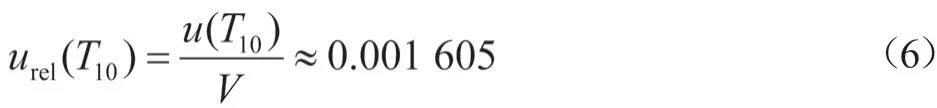
2.2.3 样品前处理引入的相对标准不确定度
依据2.2.1和2.2.2计算过程,在样品溶液配制过程中,环境温度和体积引入的不确定度分别为urel(V1)=0.005 94,urel(T1)=0.003 11。
样品溶液制备过程中产生的相对标准不确定度为:
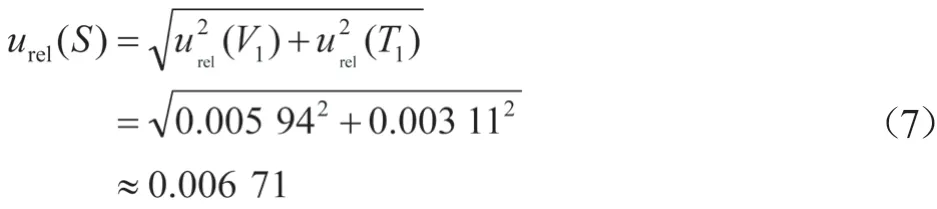
2.3 标准溶液的相对标准不确定度urel(C)
2.3.1 标准溶液引入的不确定度urel(std)
由标准品证书可知,CAP标准值的不确定度综合考虑了原料纯度定值结果、制备过程、量值核验、均匀性、稳定性等引入的不确定度分量。100 μg/mL甲醇中CAP溶液标准物质的相对误差为3%(k=2)。
其相对不确定度为:
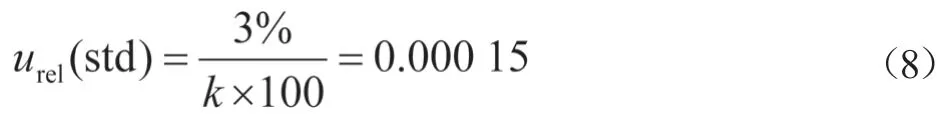
2.3.2 标准工作溶液稀释移液器引入的不确定度urel(V2)
移液器引入的不确定度由移液器校准与温度效应引入的不确定度组成。系列标准工作溶液配制所用Eppendorf移液器规格为100~1 000 μL(允差±1.0%)、20~200 μL(允差±2.0%)、2~20 μL(允差±8.0%)、10.000 mL量瓶(允差±0.020 mL),查移液器检定规程[7],测量不确定度的分析方法同2.2.1章节,不同移液体积不确定度结果分别为urel(V1000)=0.004 1、urel(V100)=0.006 1、urel(V10)=0.016 3、urel(V10量瓶)=0.000 8,使用频次分别为12、2、2、6次。由量器体积允差引入的累积相对不确定度urel(V2)=[0.004 12×12+0.006 12×2+0.016 32×2+0.000 82×6]1/2=0.028 5。
2.3.3 标准溶液制备过程温度引入的不确定度urel(T2)
在配制系列标准溶液时,移液枪头材质为聚乙烯,膨胀系数较小,受温度影响忽略不计[7]。当温度为20 ℃时,乙腈的膨胀系数为1.37×10-3/℃,分析方法同2.2.2,urel(T2)为0.001 6。
2.3.4 标准溶液的合成相对标准不确定度
在系列标准溶液配制过程中产生的相对标准不确定度urel(ρ):
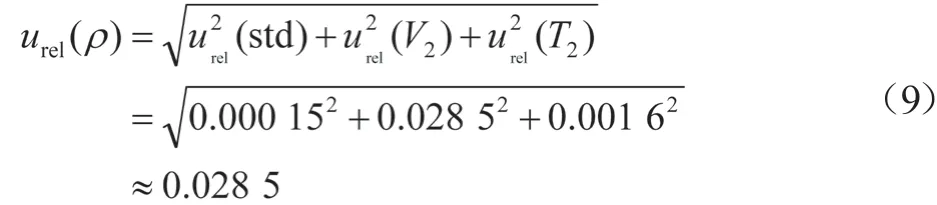
2.4 仪器校准引入的相对标准不确定度
urel(E)通常用B类不确定度评定,根据仪器说明书和校准证书,质谱和液相的最大允许误差均为1.0%[8],按均匀分布,则:
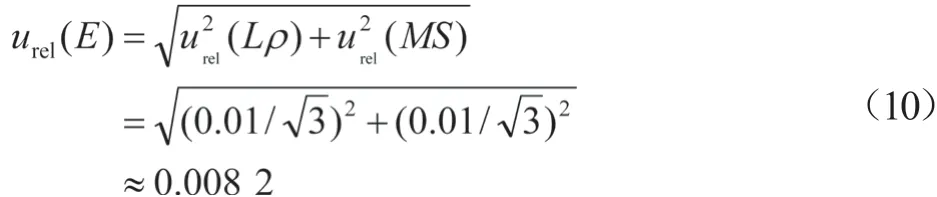
2.5 标准线性回归拟合引入的不确定度urel(q)
系列标准溶液中平均峰面积比值分别为0.081 6、0.114 0、0.169 3、0.414 2、1.036 1、2.430 3,每个质量浓度重复测定3次,测得峰面积比值以及拟合曲线y=0.490 2x-0.011 4,相关系数R=0.996 3。
测得峰面积的残余标准偏差:
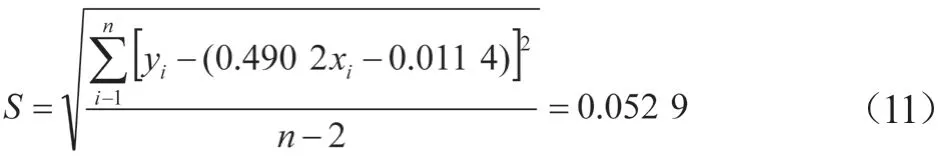
在质控样品的检测过程中,重复测量样品30次(p=30);校准曲线中CAP测定的总次数n=18;校准曲线中6个质量浓度的理论平均值ρ1=1.466 7 μg/L;测得质控样品峰面积比平均值为1.738 3,质控样品溶液中CAP质量分数X测为0.858 8 μg/kg;
由此校准曲线拟合引起的相对标准不确定度:
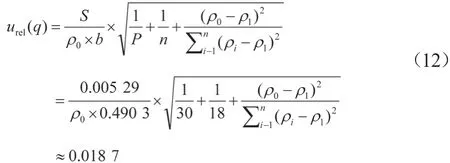
式中:ρ0为样品中CAP的质量浓度,μg/L;ρ1为标准系列溶液质量浓度的平均值,μg/L;b为拟合工作曲线的斜率;ρi为标准溶液质量浓度,μg/L;n=18为系列标准曲线总进样次数。
2.6 测量重复性引入的相对标准不确定度urel(D)
平行测定6次加标样品,平均质量分数X为0.441 μg/kg、标准偏差S(X)为0.005 7,计算得到重复测定引入的不确定度u(D)=S(X)/2.449=0.002 3和相对不确定度urel(D)=u(D)/X=0.005 3。
2.7 回收率引入的相对标准不确定度urel(R)
加标6个样品,回收率分别为85.4%、87.6%、90.2%、86.2%、89.6%、90.6%,平均回收率R为88.3%,标准偏差S(R)=0.021 9,计算得到回收率引入的不确定度u(R)=S(R)/2.449=0.008 9和相对不确定度urel(R)=u(R)/R=0.010 1。
用t检验法考察回收率R与1之间是否具有显著差异:

t大于双侧临界值t(95,5)=2.571,则回收率R与1之间具有显著差异,需要回收率R校正测定结果,则牛奶样品中CAP的质量分数为X:

2.8 标准不确定度的合成与扩展
合成不确定度等于各分量方差之和的正平方根,即合成相对标准测量不确定度公式:
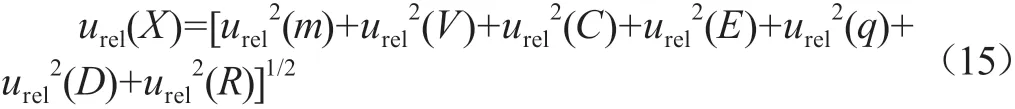
代入上述结果:urel(X)=[0.001 152+0.006 712+0.028 52+0.008 22+0.018 72+0.005 32+0.010 12]1/2=0.037 5。
通过计算,质控样中CAP的合成标准测定不确定度:
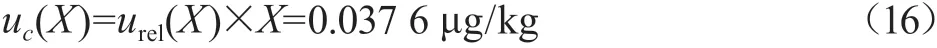
当包含因子k=2时,置信概率P为95%[9],则质控样的扩展不确定度:
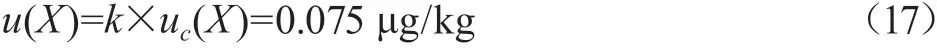
质控样品牛奶中CAP的质量分数为1.004 μg/kg,根据评定结果,牛奶中CAP质量分数测定结果X=(1.004±0.075)μg/kg,k=2。
3 讨论
本研究通过评定发现,样品前处理、基质标准溶液配制、标准曲线拟合、添加回收率、设备校准、样品称量和重复测定的不确定度分量对整体不确定度贡献较大。
在配制标准溶液的过程中,适当增大液体量取体积、使用更加精确的量取工具,可以进一步降低引入的不确定度;增加标准溶液重复测量次数、优化仪器自身数据处理系统和增加标准溶液进样量,可以降低由标准曲线拟合引入的不确定度[9-10]。通过以上试验操作,可降低各分量产生的不确定度,进一步提高检测结果的准确性和可靠性,为牛奶中CAP的质量控制提供理论依据。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国土壤与肥料(2021年5期)2021-12-02
色谱(2021年7期)2021-06-07
今日农业(2020年22期)2020-12-14
化工设计通讯(2020年10期)2020-09-17
中西医结合心血管病杂志(电子版)(2018年25期)2018-01-14
科教导刊(2017年26期)2017-11-07
中国氯碱(2016年9期)2016-11-16
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
