
新型双金属A-MnOx催化剂用于甲苯的光热催化降解
2022-02-17 13:42江善良汤颖李畅浩杨东晓刘琰赵祯霞
广西大学学报(自然科学版) 2022年6期
江善良, 汤颖, 李畅浩, 杨东晓, 刘琰, 赵祯霞
(广西大学 化学化工学院, 广西 南宁 530004)
0 引言
挥发性有机化合物(VOCs)被广泛应用于建筑材料、汽车内饰、家具合成等领域[1]。这些VOCs在应用过程中,不可避免地被释放、泄露进入大气环境中,并导致光化学烟雾、对流层臭氧、二次气溶胶等一系列环境大气污染问题[2]。据报道,VOCs具有潜在的神经毒性、生殖毒性和致癌性,即使在痕量浓度下长期接触,也会对人类生命健康构成严重威胁,尤其是甲苯、苯等苯系VOCs,因此,有效地去除大气环境中残留的VOCs至关重要,并引起了广大研究者的密切关注,目前已开发了吸附[3]、光催化[4]、热催化[5]、非热等离子体催化[6]等一系列措施解决这一问题。光催化技术被认为是一种绿色清洁的VOCs净化技术。在传统的光催化技术中,仅利用了太阳能作为光的一部分用途。为提升太阳能的利用效率,通过光热效应将太阳能转换成热能用于VOCs降解,成为一种绿色可行的解决方案[7-8]。
锰基氧化物具有价态可调、氧物种丰富、储量多、低价等特点,常常应用于热催化降解VOCs[9],并表现出优异的催化活性。同时,具有混合价态、半导体性质的锰基氧化物能够形成异质结界面[10]。这种界面的存在促进了光生电子-空穴对的有效分离,从而赋予锰基氧化物独具特色的光催化活性。此外,锰基氧化物是一种固有的深色过渡金属氧化物,能将光能转换成热能,充分提高太阳光的利用效率[11]。如今,很多工作都致力于设计具有丰富氧空位的高活性锰基氧化物,以促进对VOCs的催化活性[12-14],然而,如何在充分利用太阳光的基础上,利用光热协同效应进一步提升VOCs的催化性能,对光热催化剂的设计仍是一种挑战。
本文采用一种新型的异质原子掺杂型缺陷工程策略,利用草酸分子具有可旋转弯曲的双羧基将锰离子与A位异质原子原位螯合成键,形成锰离子与A位异质原子均匀分布的锰基草酸盐,并在临界热解温度的煅烧下,形成紧密结合的双金属锰基氧化物A-MnOx。采用SEM、EDS、XRD、Raman、TGA、XPS等表征对A-MnOx锰基催化剂的形貌、晶型、稳定性、孔道结构和表面化学性质等进行分析,并研究了A-MnOx锰基催化剂对甲苯光热催化性能及机理。本文还通过不同条件下的O2-TPD、H2-TPR等表征揭示了光热协同催化的机理,为苯系VOCs高效净化提供了一种可行的途径。
1 实验
1.1 材料
醋酸锰(AR,美国Sigma-Aldrich公司);无水草酸、硝酸铈、硝酸钴(AR,上海阿拉丁生化科技股份公司);硝酸铜、硝酸镍(AR,国药集团化学试剂公司);硝酸铁(AR,西陇科学股份有限公司)。其他所用试剂均为商业购买,不需要进行纯化即可直接使用。
1.2 不同异质原子掺杂A-MnOx的制备
将 30 mmol的醋酸锰添加到150 mL超纯水中超声30 min,并在转速800 r/min下持续搅拌,直至醋酸锰完全溶解到溶液中,形成均相溶液。
将等摩尔的草酸加入到上述溶液中,并在剧烈搅拌下反应50 min。
经过抽滤装置将固液分离,并放置到80 ℃的烘箱中干燥过夜,获得淡粉色的草酸锰颗粒,命名为MnOA。
将得到的MnOA置于马弗炉中煅烧(煅烧条件为空气气氛,升温速率为5 ℃/min,煅烧温度为300 ℃),并维持6 h。获得的锰基催化剂命名为MnOx。
双金属A-MnOx系列材料的制备过程与单金属MnOx相似。以Cu-MnOx为例,将1.5 mmol硝酸铜和28.5 mmol醋酸锰加入到150 mL超纯水中,超声搅拌溶解,然后加入30 mmol的草酸,在剧烈搅拌中持续反应50 min。通过过滤获得淡蓝绿色的锰铜草酸盐,命名为Cu-MnOA。经过300 ℃煅烧后,获得锰铜双金属氧化物,命名为Cu-MnOx。
同样地,将Cu盐替换成不同金属的硝酸盐(Fe、Ni、Ce、Co),分别获得Fe-MnOx、Ni-MnOx、Ce-MnOx、Co-MnOx。
1.3 仪器表征
采用X射线衍射仪(XRD,SMARTLAB3KW型, 日本理学公司)、拉曼光谱仪(Raman, LabRAM HR Evolution型, HORIBA Jobin Yvon)对材料的晶体结构和化学组成进行表征分析。采用扫描电子显微镜(SEM, Hitachi SU8220型, 日本日立公司)分析表征材料的微观表面形态和元素分布。采用比表面积和孔径分析仪(ASAP 2460型, 美国麦克有限公司)测试材料的比表面积和孔径结构。通过热重分析仪(TGA,TGA/DSC3+/1100LF型, 梅特勒-托利多有限公司)表征材料的热稳定性能。X射线光电子能谱(XPS, ESCALAB 250XI+型, 美国赛默飞科技公司)分析材料表面元素组成和化学价态。
1.4 甲苯催化降解测试
将100 mg催化剂装载到石英管中,通过体积流量为50 mL/min的干空气将750 mg/m3的甲苯蒸气携带到反应器中,采用装有可见光滤波片的氙灯作为光源,光强为140 mW/cm2,进行光热催化反应。反应产物通过配备由FID和TCD检测器的气相色谱仪(GC7890B型,美国安捷伦科技有限公司)实时监测。同样,在甲苯吸附平衡后,采用电加热的方式,以1 ℃/min的升温速率,进行热催化反应。
2 结果与讨论
2.1 催化剂的微观形貌分析
采用SEM和EDS对不同异质原子掺杂的A-MnOx的微观形貌进行表征,结果如图1所示。
从图可见,原始MnOx呈现为致密的类面包状团聚体结构,且具有较大的晶体尺寸(约4 μm),不利于MnOx催化位点的有效暴露和VOCs的扩散传质反应[图1(a)]。图1(b-f)分别对应于Fe-MnOx、Ni-MnOx、Ce-MnOx、Co-MnOx和Cu-MnOx,从中可以发现,相比较于原始MnOx,其晶体颗粒明显变小,表明异质原子的原位修饰在一定程度上能抑制MnOx晶面的结晶生长并产生晶体缺陷[15-16]。A-MnOx不同的晶体形貌说明不同的异质原子掺杂产生不同程度的晶体缺陷。具体来说,Fe-MnOx和Ni-MnOx的晶体尺寸仅减小至约1.5 μm,且其表面的Fe、Ni离子分布较少,表明在Mn草酸螯合配位过程中,Fe和Ni的竞争配位能力较弱,导致只有少量Fe和Ni参与配位成功,并产生少量的晶体缺陷。相比较之下,Ce-MnOx表现为片状无序堆叠结构,说明Ce的引入影响了草酸锰晶核的生长[17],使得Mn与草酸在二维平面上螯合配位,这种片层结构有利于暴露更多的活性位点,促进VOCs在其上的扩散及反应。值得注意的是,Co-MnOx和Cu-MnOx均为表面结构塌陷并伴随大量裂缝的块状破碎体结构,表明Co和Cu与草酸分子之间存在较好的亲和力,能与Mn形成竞争配位,在抑制MnOx结晶的同时进一步产生丰富的晶体缺陷。根据相应的EDS谱图发现,Co-MnOx和Cu-MnOx的异质原子掺杂均能成功诱使紧密双金属界面的形成,丰富的晶体缺陷有利于暴露更多的催化位点,进一步强化VOCs的催化氧化性能。

(a) MnOx

(b) Fe-MnOx

(c) Ni-MnOx

(d) Ce-MnOx

(e) Co-MnOx
图2(a)为A-MnOA煅烧热解后形成的双金属锰基氧化物A-MnOx的PXRD谱图。从中可以看到,由无金属掺杂MnOA衍生的MnOx存在较弱的晶体衍射峰,表明其草酸锰结构在低温煅烧的过程中已全部分解,并形成具有晶体缺陷的锰基氧化物。通过与标准PDF卡片比对发现,MnOx在37.311°〔(-111)晶面〕、47.987°〔(202)晶面〕和65.623°〔(020)晶面〕处的衍射峰与δ-MnO2(JCPDS PDF#80-1098)相一致[18]。经过异质原子的原位修饰后,形成的双金属A-MnOx在18.013°〔(101)晶面〕、30.382°〔(103)晶面〕、36.083°〔(211)晶面〕处出现新的衍射峰,并与Mn3O4(JCPDS PDF#80-0382)相匹配[19],表明异质原子的原位掺杂能抑制MnOx的结晶并诱导丰富晶体缺陷的产生,从而使Mn的表面价态降低并形成δ-MnO2/Mn3O4多晶界面[20]。值得注意的是,XRD谱图中观察不到异质原子相应氧化物的特征峰,再次证明异质原子能与Mn形成紧密结合的双金属界面,而不是简单混合的金属氧化物。这种嵌入到MnOx晶格中的异质原子更有利于内部电子的转移,进而赋予A-MnOx优异的催化氧化能力。利用N2等温吸附-脱附曲线对双金属A-MnOx的比表面积和孔隙结构进行研究,结果如图2(b)和表1所示。

(b) 氮气等温吸附-脱附曲线

表1 双金属锰基氧化物的结构参数及表面元素质量分数Tab.1 Textural property and surface element distribution of bimetallic Mn-based oxides
所有样品都呈现为典型的IV等温吸脱附曲线,其在低相对压力(p/p0< 0.1)下存在N2的快速吸附,这是典型的微孔填充特征。值得注意的是,这些样品在p/p0> 0.4时,N2脱附量大于吸附量,并表现出明显的H3型回滞环,意味着材料中存在着不规则的裂缝型孔道结构。这可归因于草酸盐在热分解的过程中,CO2的原位生成并溢出所致。根据表1可以观察到,异质原子原位修饰后,双金属A-MnOx的比表面积是原始MnOx的1.33~2.26倍,由小到大的顺序为MnOx(125.4 m2/g)、 Fe-MnOx(167.2 m2/g)、Ni-MnOx(189.1 m2/g)、Ce-MnOx(201.5 m2/g)、Co-MnOx(224.3 m2/g)、Cu-MnOx(283.0 m2/g)。有趣的是,与原始MnOx相比,双金属A-MnOx的总孔容明显增大,证明异质原子的掺杂能抑制MnOx结晶并产生更大的缺陷孔,这与SEM表征相呼应。同时,Cu-MnOx的介孔孔容显著提升,是MnOx和其他异质原子掺杂A-MnOx的1.31~1.89倍,进一步证明Cu具有更强的草酸螯合竞争配位能力,能更好地嵌入MnOx晶格并形成紧密的双金属界面,进而产生更多的晶体缺陷,暴露出更大的比表面积和更多缺陷孔。

图3 双金属锰基氧化物的XPS谱图Fig.3 XPS spectra of bimetallic Mn-based oxides
通过XPS进一步对双金属A-MnOx的元素组成和表面化学性质进行探究,结果如图3所示。
可见,所有的样品均有Mn、O和相应的异质原子A组成,其相应的A与Mn原子比由小到大依次为MnOx(0)、Fe-MnOx(3.87%)、Ni-MnOx(4.82%)、Ce-MnOx(6.17%)、Co-MnOx(6.46%)、Cu-MnOx(6.51%)。与此同时,这些样品的O与Mn原子比呈现为负相关关系,由小到大顺序为Cu-MnOx(1.96)、Co-MnOx(2.00)、Ni-MnOx(2.04)、Ce-MnOx(2.05)、Fe-MnOx(2.16)、MnOx(2.22)。从中可以发现,在相同掺杂量的情况下,异质原子的表面负载量存在差异,并且A-MnOx中的A与Mn原子比越大,就能更有效地抑制MnOx的结晶生长并产生晶体缺陷和晶格畸变,进而伴随着更多的不饱和金属氧键的生成和丰富氧空位的出现。其中,Cu-MnOx具有较大的A与Mn原子比和最小的O与Mn原子比,说明Cu异质原子与MnOx结合良好,更容易嵌入MnOx晶格并产生更多的晶体缺陷和丰富氧空位。
图4为双金属A-MnOx的高分辨Mn 2p和O 1s XPS谱图。
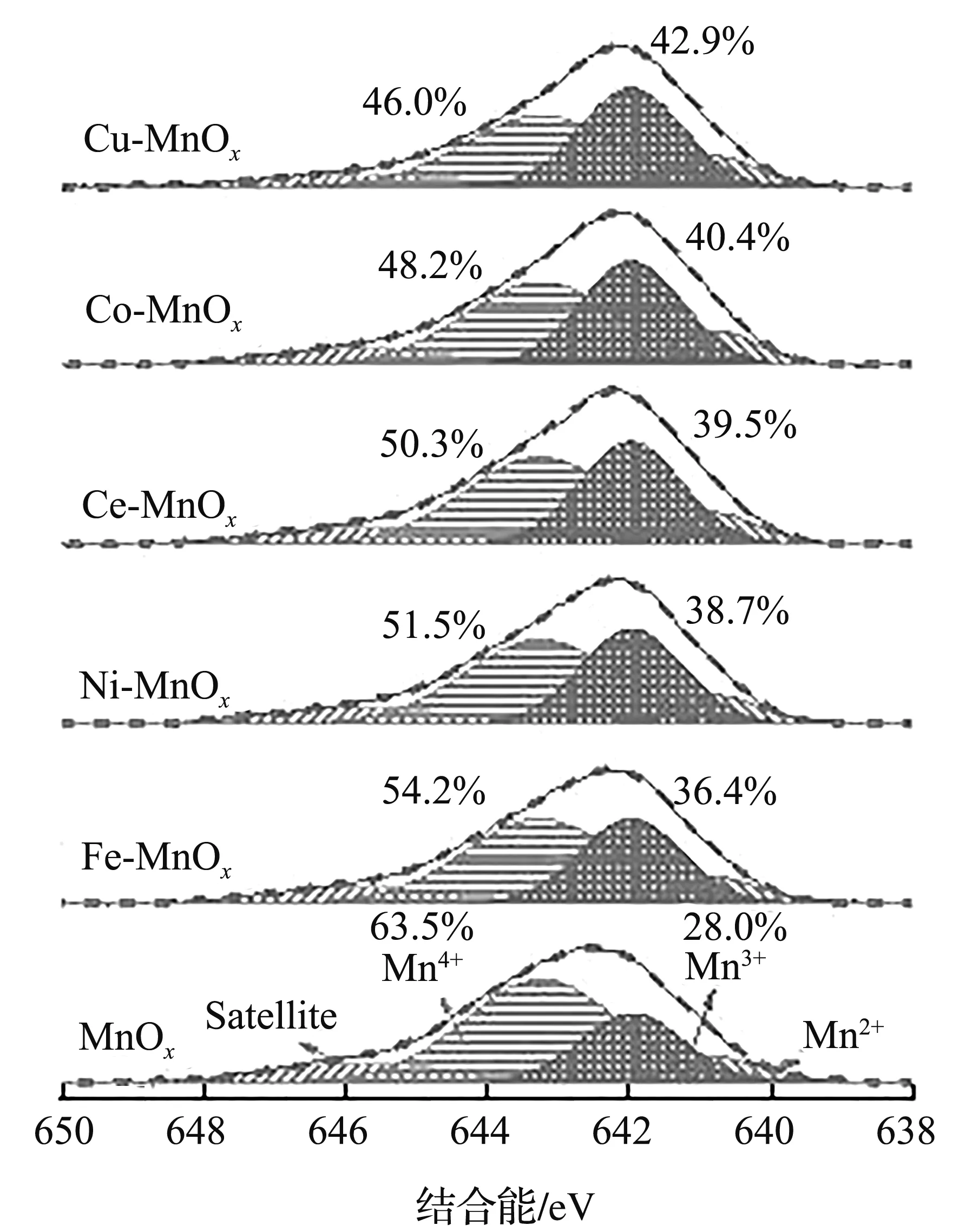
(a) Mn 2p
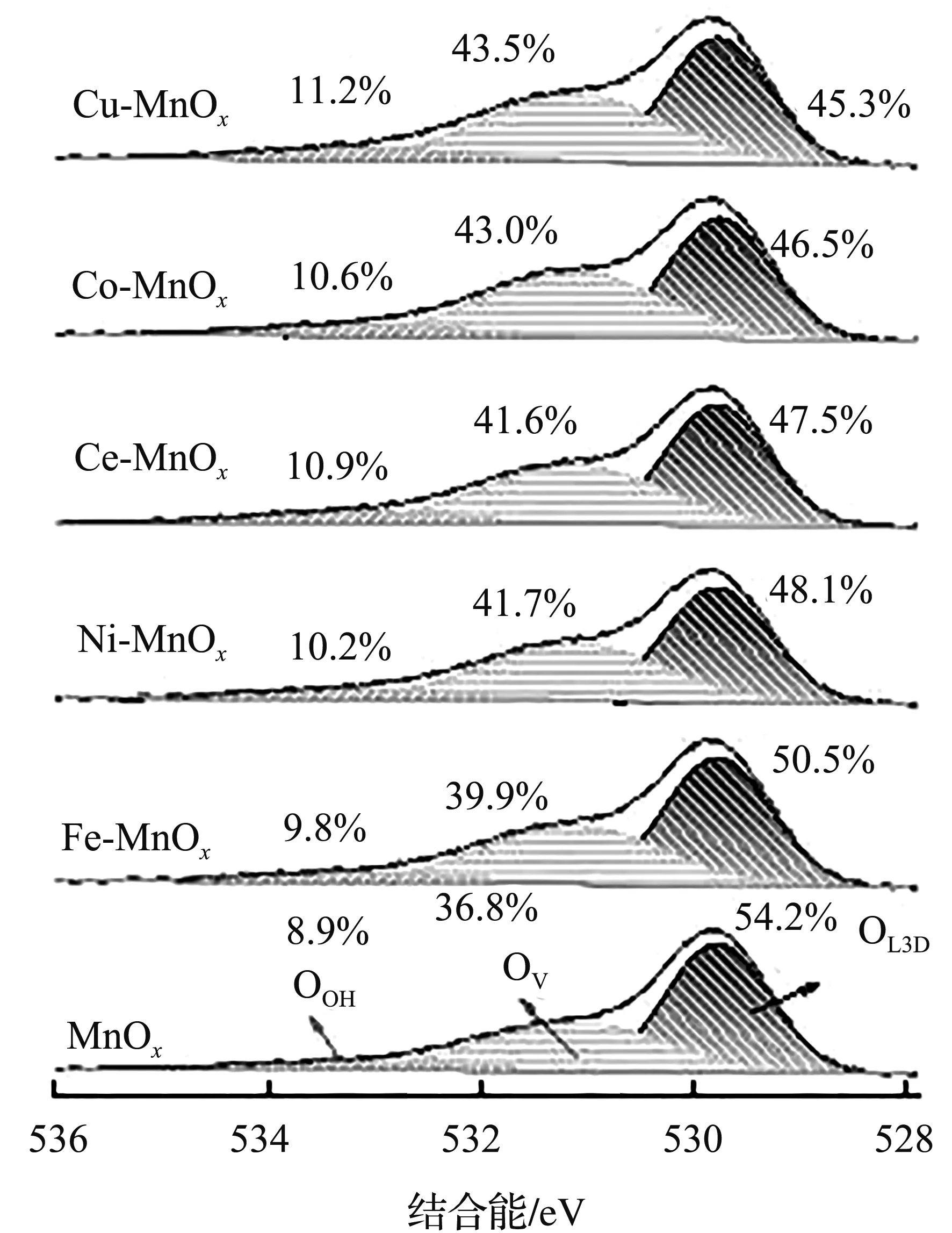
(b) O 1s
所有样品在结合能为640.8、641.9、643.3、646.2 eV处存在4个峰,分别对应着Mn2+、Mn3+、Mn4+和卫星峰[14]。据文献[15,18]报道,氧空位在VOCs的低温催化中起重要作用。有趣的是,随着异质原子的引入,MnOx中的Mn3+与Mn4+离子比(44.1%)有逐渐增加的趋势,表明异质原子的原位掺杂会诱导产生更多的晶体缺陷,并促进更多的δ-MnO2晶体转变成Mn3O4晶体。相比较之下,Cu-MnOx的Mn3+与Mn4+离子比最大(93.3%),分别是原始MnOx和其他异质原子掺杂A-MnOx的2.11倍和1.11~1.39倍,进一步证实Cu能更好地嵌入MnOx晶格并促进缺陷多晶界面的生成。从图4(b)的O 1s XPS谱图可以看到,这些样品的结合能在533.2、531.1、529.8 eV处出峰,分别对应于表面吸附水(OOH)、表面氧空位(OV)和体相晶格氧(OLatt)[14-15,18]。总所周知,OV含量越高,气相O2越容易吸附和活化成具有低温催化活性的氧物种[18]。结合Mn 2p分析发现,异质原子的原位掺杂能诱使其表面出现更多的不饱和金属氧键,丰富的界面缺陷使其OOH和OV含量增加,而OLatt含量减少。通过对O 1s进行反卷积计算发现,随着异质原子的引入,样品的(OOH+ OV)与OLatt离子比呈规律变化,由小到大的顺序为MnOx(0.84)、Fe-MnOx(0.98)、Ni-MnOx(1.08)、Ce-MnOx(1.11)、Co-MnOx(1.15)、Cu-MnOx(1.21),表明异质原子的原位修饰在促进缺陷结构形成的同时,还有利于丰富氧空位的生成。Cu的引入能更好地与MnOx晶格结合,使Cu-MnOx产生更多的氧空位并具有更强的催化氧化能力。
2.2 双金属锰基催化剂A-MnOx对甲苯的光热降解性能研究
图5为双金属锰基氧化物A-MnOx的甲苯降解曲线。

(a) 热催化性能

(b) 光热催化性能
一般来说,考察催化剂的催化活性主要是看其低温催化活性、高温稳定性,即转化率为10%的起燃温度T10和完全转化温度T90。为了能更好地探究光衍生光热催化的优越性,本文探究了在空速为30 000 mL/(g·h)、甲苯浓度为750 mg/m3条件下催化剂的热催化性能,如图5(a)所示。在进行热催化测试前,所有样品均进行300 ℃的干空气下活化1 h。从图中可以看出,在空速为30 000 mL/(g·h)、甲苯浓度为750 mg/m3条件下,原始MnOx在150 ℃前其甲苯转化效率随温度的增加而逐渐提升,随后开始下降,而当温度达到195.8 ℃后,其甲苯转化效率开始快速提高,完全转化温度T90为246.7 ℃。第一阶段甲苯催化活性的提升是由MnOx表面吸附氧等低温活性物种引起的催化氧化。随着MnOx表面吸附氧物种的逐渐消耗却没有被及时补充,导致其活性开始逐步下降。当反应温度大于190 ℃,具有低温催化活性的氧空位获得足够的能量,重新对吸附氧物种进行活化,并且MnOx中的晶格氧开始在热作用下激活,形成具有较强氧化能力的活性氧[O],从而促进了第二阶段甲苯催化效率的快速提升。相比较之下,异质原子掺杂的A-MnOx均表现出高于原始MnOx的表面活性氧含量,且其热激活氧空位所需的温度明显降低,其中Cu-MnOx表现最优,起燃温度T10仅为172.3 ℃,比原始MnOx降低了23.5 ℃,说明异质原子的引入产生的丰富晶体缺陷能有效促进A-MnOx对氧物种的吸附,同时形成的丰富氧空位能有效捕获并活化氧物种,显著提升A-MnOx的低温催化氧化能力。Cu-MnOx具有丰富的表面缺陷和微介孔结构,能暴露出更多的催化位点,同时强化甲苯的传质,从而在229.6 ℃就实现约90%的甲苯转化率,明显优于原始MnOx和其他异质原子掺杂的A-MnOx。
图5(b)为不同锰基氧化物在空速为30 000 mL/(g·h)、甲苯浓度为750 mg/m3、光强为140 mW/cm2条件下的光衍生光热催化甲苯降解性能。可见,所有样品在前10 min内均表现出快速的甲苯催化氧化和CO2生成,达到最高值后出现不同程度的下降。其中,原始MnOx在光热催化130 min后,其甲苯转化效率为35.7%,相应的CO2产率为25.3%。随着不同异质原子的引入,A-MnOx的甲苯催化氧化能力出现不同程度地提升,其甲苯转化效率由低到高的顺序为MnOx(35.7%)、Fe-MnOx(56.7%)、Ni-MnOx(68.3%)、Ce-MnOx(71.6%)、Co-MnOx(83.1%)、Cu-MnOx(94.5%),其相应的CO2产率呈正相关关系递增,由低到高的顺序为MnOx(25.3%)、Fe-MnOx(46.4%)、Ni-MnOx(58.0%)、Ce-MnOx(59.9%)、Co-MnOx(73.2%)、Cu-MnOx(82.8%)。值得注意的是,在相同条件下,Cu-MnOx的反应平衡温度仅为157.1 ℃即可实现远高于90%的甲苯转化率,比热催化条件降低了72.5 ℃,并且Cu-MnOx的甲苯降解效率是原始MnOx和其他异质原子掺杂A-MnOx的2.65倍和1.14~1.67倍,并具有更好的甲苯矿化效果,表明Cu的原位修饰能诱导Cu-MnOx产生更多的晶体缺陷和暴露更多的催化位点,进一步使Cu-MnOx具有优异的光热催化活性。
2.3 光热催化降解机理
图6为双金属氧化物A-MnOx的EIS曲线和O2-TPD曲线。

(a) EIS曲线

(b) O2-TPD曲线
为了深入阐明异质原子掺杂促进催化活性提升的原因,对A-MnOx的电子传输和氧迁移能力进行分析。电子转移性能是影响光热催化活性的关键因素之一,因此本工作采用EIS对不同锰基氧化物的导电性能进行研究。测试得到的锰基氧化物的Nyquist曲线如图6(a)所示,在1.0 ~100.0×103Hz的频率范围内扫描出半圆,其半径反映了电极的电荷转移电阻值RCT。所有样品的RCT值由小到大的排序依次为Cu-MnOx(212.4 Ω)、Co-MnOx(219.1 Ω)、Ce-MnOx(229.0 Ω)、Ni-MnOx(258.3 Ω)、Fe-MnOx(268.2 Ω)、MnOx(311.9 Ω)。可见,原始MnOx具有最大的RCT值,随着异质原子的引入,其电子转移能力得到明显增强,证明了异质原子与Mn形成紧密结合的双金属界面促进了其金属到金属之间的电荷转移[12-13,21]。其中,Cu-MnOx具有最优的导电性能,表明Cu能更好地嵌入至MnOx的晶格内,并诱导其产生大量的晶体缺陷和丰富的氧空位,进一步提高其电导率。
为了研究金属掺杂对氧物种活性和迁移的影响,分别对原始MnOx、Fe-MnOx、Ni-MnOx、Ce-MnOx、Co-MnOx和Cu-MnOx催化剂进行O2-TPD测试,结果如图6(b)所示。在开始测试前,所有样品在300 ℃条件下,以体温流量为30 mL/min的氦气流预处理1 h,以消除物理吸附杂质的影响。值得注意的是,活性氧存在3个解吸峰:① 位于表面氧空位的化学吸附氧(375~450 ℃,标记为OV);② 不饱和金属氧键释放的表面晶格氧(450~565 ℃,标记为OSurf);③ 高温处理过程中从金属氧化物的价态转变中分离出来的体相晶格氧(675~825 ℃,标记为OLatt)[5,7,11,18]。可见,原始MnOx主要以OLatt为主,随着异质原子的引入,其逐渐转变成以OV和OSurf为主。同时,OV和OSurf的脱附峰明显向低温方向偏移。这一结果表明,异质原子的原位掺杂将导致A-MnOx产生含有大量不饱和金属氧键的晶体缺陷结构,并促使活性氧物种相催化剂表面的有效迁移,更有利于低温下活性氧的释放。值得注意的是,Cu-MnOx中OV和OSurf的解吸峰强度最高,并进一步向低温方向偏移,表明Cu能更好地嵌入MnOx的晶格并对MnOx的MnO6八面体结构产生影响,产生更多的晶体缺陷的同时伴随着更多的不饱和金属氧键,这与XPS分析相吻合。此外,这一现象更直观地证明了Cu-MnOx具有丰富的氧空位和强烈的吸附氧性能,独特的多晶界面赋予其更好的低温活化氧物种能力,进而使其对甲苯具有优异的光热催化氧化活性。
在光照和黑暗条件下分别测定Cu-MnOx的O2-TPD和H2-TPR,以研究其光热协同催化机理,结果如图7所示。

(a) O2-TPD
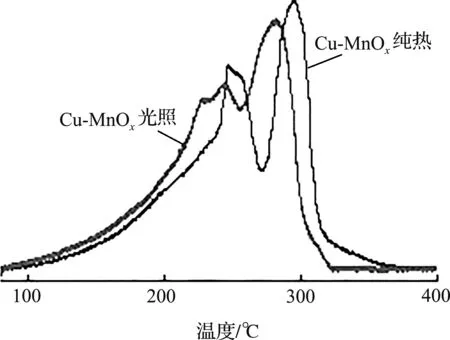
(b) H2-TPR
同样地,首先将Cu-MnOx的物理吸附杂质去除,以便后续研究。根据图7(a)的O2-TPD发现,在光照条件下,Cu-MnOx的氧空位吸附活性氧和晶格氧的强度都比暗条件下有所提高,且明显向低温偏移。这一结果表明,光与光驱动产热的协同作用可以强化更多氧的解吸,并有效降低Cu-MnOx中晶格氧的光活化能垒[22]。同时,Cu的原位掺杂能诱导Cu-MnOx产生大量晶体缺陷和丰富的氧空位,并伴随着大量的不饱和金属氧键,更有利于氧的解吸和更活跃的表面晶格氧的生成。在光热协同下的H2-TPR测试中也出现了类似的结果[图7(b)],Cu-MnOx的峰在光照下依旧有明显的向低温方向偏移,进一步说明Cu的原位修饰产生了更多的缺陷结构和不饱和金属氧键,从而产生了更多的活性氧物种,光热协同效应能有效增强其低温还原性,并赋予Cu-MnOx更优异的光热催化活性。
3 结语
本文采用异质原子原位掺杂策略,将不同的金属离子引入到草酸锰的合成体系中,成功制备出富含氧空位的双金属锰基氧化物A-MnOx,并将其应用到甲苯的光热催化降解中,实现了能源减排和环境治理的功效。研究了双金属锰基氧化物A-MnOx的形貌、晶型、稳定性、孔道结构和表面化学性质,对甲苯的光热协同催化机理进行讨论,得出以下结论:①异质原子掺杂可以增大催化剂的缺陷浓度,从而提高氧空位含量;②异质原子掺杂通过增强内部电子的流动,促进了催化剂的导电和氧化还原能力;③光热协同作用有利于活性氧物种的活化和迁移,从而加强催化剂的催化活性。通过异质原子掺杂获得的Cu-MnOx具有丰富的氧空位和优异的催化活性,其甲苯光热降解能力为别是原始MnOx和其他A-MnOx的1.14~2.65倍,并具有长时间的催化稳定性,为苯系VOCs的降解提供了一种高效可行的途径。
猜你喜欢
化工管理(2022年13期)2022-12-02
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
陶瓷学报(2019年5期)2019-01-12
能源(2017年9期)2017-10-18
制冷技术(2016年3期)2016-12-01
能源(2016年10期)2016-02-28
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
太阳能(2015年12期)2015-04-12
读者欣赏(2014年6期)2014-07-03
