
离子液体在镧系锕系元素萃取分离应用中的理论计算研究进展
2022-03-14 06:50张宇生孙涛祥
核化学与放射化学 2022年1期
王 强,张宇生,孙涛祥
清华大学 核能与新能源技术研究院,北京 100084


图1 离子液体中常用的阴阳离子Fig.1 Common cations and anions of ionic liquids

在目前已有的离子液体体系萃取镧系锕系元素的研究中,部分理论计算工作采用分子动力学模拟和量子化学计算手段对离子液体在萃取分离中与金属离子的相互作用进行了研究。夏苗仁等[6]总结了近年来镧系和锕系元素溶液动力学的研究进展,文中对离子液体萃取镧系锕系元素进行了简单介绍,认为静电相互作用决定金属离子的溶剂化结构及其动力学。为了更深入地认识离子液体与镧系锕系金属离子的相互作用,本文对镧系锕系金属离子和配合物在离子液体中与离子液体的阴、阳离子相互作用的分子动力学模拟和量子力学计算进行了综述,明确离子液体的阴、阳离子与镧系锕系金属离子的配位作用以及镧系锕系金属离子和配合物在离子液体中的溶剂化特征,以期对离子液体体系萃取镧系锕系金属离子的化学机理有更深入的认识。
1 研究方法概述
1.1 分子动力学模拟
分子动力学模拟(molecular dynamics, MD)是利用牛顿物理力学方程来计算一个多组分体系平衡和传递性质的分子模拟方法,具有简单、高效、较强的可行性以及能够从原子层面给出体系的微观演变过程等优点,能直观地展示实验现象发生的机理与规律。
在分子动力学模拟中,系统的能量是所有粒子位置的函数,在确定粒子坐标后,能够计算出体系的能量;而体系的总能量对粒子位置的负梯度值是该粒子所受到的作用力,根据经典力学的运动方程能够求解粒子的速度,同时需要考虑温度等其他因素的影响,对计算的速度进行优化后获得粒子的坐标,在确定新坐标的条件下重复循环并持续演化,直到达到模拟设定的时间。分子动力学模拟过程中选择合适的力场参数至关重要。力场处理代表系统中原子间相互的势能函数,保证了分子动力学模拟的稳定性和有效性[7]。力场可分为极化力场和非极化力场。非极化力场由于函数形式简单、表达式容易理解、计算量较小等优点在物理化学模拟中得到广泛应用。但非极化力场中除力场参数外,电荷与原子之间相互作用是不能发生改变的,对于含有较高电荷粒子带来显著极化效应的体系,会引起较大误差。因此在非极化力场的基础上发展出了极化力场,能够改进粒子间的相互作用,然而由于极化力场采用的三种模型(点偶极模型、浮动电荷模型以及壳模型)计算量庞大,因此其仍处于发展阶段,仅用于特殊体系中。力场表示能量依赖于系统中原子坐标位置的数学表达式,常用的力场形式表示为:
(1)
式中:前四项为成键项,第五项为范德华项,最后一项为静电项。kb、kθ、kn、ke以及Vimp分别代表键伸缩、角弯曲、二面角扭曲项的力常数、库伦常数以及赝扭曲势能的函数;b0、n和δ分别代表平衡的键长、二面角的旋转对称性相关的旋转多重度和相因子;qi和qj分别是i、j粒子上的电荷数;b、θ、φ以及rij分别代表原子-原子组合的键长、键角、二面角以及两个粒子间的距离;εij和σij通常通过特定组合规则计算得到;LJ表示Lennard-Jones势参数,描述非键分子或原子之间的相互作用。典型的力场描述了键角、键长、扭力、赝扭曲势能和非键项(包括范德华力和静电相互作用)的时间演变。用于分子动力学模拟的工具包括GROMACS、AMBER、LAMMPS、QUANTUM等[8-9]。
1.2 密度泛函理论概述
量子化学是基于量子力学的原理和方法研究分子的结构、性能以及结构和性能之间的关系,其核心问题是求解原子-分子体系的Schrödinger方程。量子化学的计算方法主要包括从头计算法、半经验方法以及密度泛函理论。密度泛函理论(density functional theory, DFT)是一种研究物质结构统计性的理论,既可以用于研究微观结构,也可以用于研究宏观结构,前者是量子力学的密度泛函理论,后者是统计力学的密度泛函理论。在密度泛函理论中,体系的能量和其他性质可以表达为电子密度ρ(r)的泛函,因此计算时只需要对电子密度分布函数进行变分,极大地减少了计算量,成为量子化学计算的主流方法之一[10]。
密度泛函理论实质在于计算体系的电子密度分布和基态能量,具体计算方法的核心是Hohenberg-Kohn定理和Kohn-Sham方程。Hohenberg-Kohn第一定理指出体系的基态能量仅仅是电子密度的泛函,Hohenberg-Kohn第二定理指出能获得的最低能量的电子密度即为基态电子密度。Kohn和Sham在Hohenberg-Kohn定理的基础上将动能项、外势能项、相互作用项进行了近似和简化后得到Kohn-Sham方程:
(2)

镧系锕系元素均为重元素,在其原子核附近的内层电子运动速度快,相对论效应显著,导致重元素在原子基态、电离势、电子亲和势和原子半径等方面有不同于轻元素的特点,并影响其化学性质。因此镧系和锕系元素理论化学计算时需要考虑相对论效应,密度泛函理论中对相对论的考虑主要有全电子法和有效核实势法。核实势法(effective core potentials, ECPs)的主要物理思想是将原子的内层电子连同原子核构成一个核实,核实对外层电子的作用以某种合适的有效核实势来表达,与全电子法相比可大大降低计算量。ECPs主要有模型势(model potentials, MPs)和赝势(pseudo potentials, PPs)两个分支,后者的配套基组通常更小,因此计算量更低,应用更广泛[12]。
2 离子液体体系萃取分离镧系元素的理论计算研究
稀土元素用途广泛,来源稀缺,因此需要考虑从工业废弃物中回收稀土元素,其中液-液萃取分离技术非常关键。液-液萃取技术另一个与稀土相关的重要应用是乏燃料后处理分离裂片元素[13]。离子液体的诸多理化特性使其在镧系元素的萃取分离方面表现出一定的优势,而这种优势在一定程度上来源于离子液体中特殊的配位环境。


图2 [BMI][PF6]和[EMI][TCA]中阴、阳离子的第一溶剂化壳层示意图[15]Fig.2 Snapshots of the first solvation shell of anions and cations in [BMI][PF6] and [EMI][TCA][15]

左图:第一溶剂化壳层;中间图:第一、二溶剂化壳层;右图:第一、二、三溶剂化壳层图3 潮湿[BMI][PF6]中Eu3+的溶剂化壳层示意图[16]Fig.3 Snapshots of the solvation shells of Eu3+ in the humid [BMI][PF6][16]



洋红色为单齿配位,蓝色为双齿配位图4 在9倍(a)和10倍(b)硝酸盐配位的情况下Ce3+的第一溶剂化壳层示意图[18]Fig.4 Snapshots of the first solvation shell of Ce3+ in the case of the 9-fold(a) and 10-fold(b) nitrate coordination[18]

镧系金属离子与离子液体中阴、阳离子发生相互作用时容易形成以镧系金属离子为核心的洋葱式结构:镧系金属离子首先被离子液体阴离子包围,外围又被离子液体阳离子所环绕,整体会以更加稳定的配位形式存在。在实际萃取体系中,体系的萃取能力主要依赖于萃取剂对镧系金属离子的配位作用,离子液体的阴、阳离子是否会与镧系金属离子-萃取剂的配合物发生配位作用,以及这种配位作用是否是配合物稳定结构、是否是提升萃取能力的关键因素值得深入研究。
3 离子液体体系萃取分离锕系元素的理论计算研究
锕系元素,尤其铀作为核燃料循环中最重要的元素,在离子液体体系的萃取分离过程中备受关注。Wipff课题组[20-23]不仅利用分子动力学模拟对镧系元素进行大量研究,而且对U(Ⅵ)在离子液体中与离子液体的阴阳离子的相互作用也进行了充分研究。


左图:第一溶剂化壳层;右图:第二溶剂化壳层图5 C4mimNTf2中铀酰配合物的溶剂化壳层示意图[20]Fig.5 Snapshots of the solvation shells of uranyl complexes in C4mimNTf2[20]
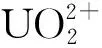


图6 [BMI][NTf2]和[MeBu3N][NTf2]中铀酰复合物的溶剂化壳层示意图[21]Fig.6 Snapshots of the solvation shells of uranyl complexes in [BMI][NTf2] and [MeBu3N][NTf2][21]



左图:MeBu3N+,右图:图7 [MeBu3N][NTf2]中配合物的第一溶剂化壳层示意图[24]Fig.7 Snapshots of the first solvation shells of complexes in [MeBu3N][NTf2][24]

铀酰离子与离子液体中阴离子发生相互作用同样能够形成与镧系金属离子类似的洋葱式溶剂化结构,即铀酰离子首先被离子液体阴离子包围,外围又被离子液体阳离子所环绕。当离子液体中含有水分子时,铀酰离子的第一溶剂化壳层会由离子液体阴离子转化为水分子,而离子液体的阴离子与铀酰离子发生外配位作用。在萃取剂存在时,离子液体的阴离子会与锕系金属离子-萃取剂的配合物发生配位作用,但水分子的存在会破坏这种配位作用。

(a)——Am3+周围的第一溶剂化壳层,共有9个配位键;(b)——仅显示Am3+∶L配合物,隐藏阴离子;(c)——仅显示Am3+周围的阴离子,隐藏L2+配体图9 [BMIM][NTf2]中L2+配体在咪唑鎓基团周围次级溶剂化壳层结构示意图[27]Fig.9 Snapshot of the secondary solvation environments around the imidazolium groups of the L2+ ligand in the [BMIM][NTf2][27]
4 总结与展望

在实际应用中,尤其是在乏燃料后处理等工程化应用中,萃取体系的应用前景不仅取决于体系的萃取性能,也与其他各种因素密切相关。具体到离子液体萃取体系,离子液体自身的密度比水大,因此在萃取设备设计、已有设备运行方面需要考虑反相的问题;离子液体的黏度较大,设备运行过程存在一定的水力学问题和传质问题。以上问题制约着离子液体萃取体系的工程应用,也促使研究者对离子液体的物化性质进行进一步的优化和改进。
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
北京航空航天大学学报(2022年7期)2022-08-06
作文周刊·小学一年级版(2022年24期)2022-06-18
小学阅读指南·低年级版(2022年5期)2022-05-09
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
黑龙江大学自然科学学报(2022年1期)2022-03-29
当代水产(2021年10期)2022-01-12
中学生数理化·八年级物理人教版(2021年4期)2021-07-22
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
