
红芪药材、饮片、标准汤剂与配方颗粒的高效液相色谱指纹图谱相关性研究
2022-03-30 02:05毛小文祁梅顾志荣马转霞葛斌吕鑫张锐甘肃中医药大学药学院兰州730000甘肃省人民医院药剂科兰州730000
中南药学 2022年2期
毛小文,祁梅,顾志荣*,马转霞,葛斌*,吕鑫,张锐(. 甘肃中医药大学药学院,兰州 730000;.甘肃省人民医院药剂科,兰州 730000)
红芪为豆科植物多序岩黄芪(Hedysarum polybotrys
Hand.-Mazz.)的干燥根,味甘,性微温,归肺、脾经,有补气固表、利尿、排脓托毒、敛疮生肌等功效,临床上常用于治疗气虚乏力、中气下陷、表虚自汗、气虚水肿、痈疽难溃等症。红芪为甘肃十大陇药之一,大面积栽培于陇南武都东南部山区地带。现代药理研究表明,红芪中含多糖、黄酮、皂苷等多种生物活性成分,具有提高免疫力、抗病毒、抗氧化、抗肿瘤等广泛的药理作用。以符合炮制标准的红芪饮片为原料,通过现代提取、浓缩、喷雾干燥、制粒等技术制备成红芪配方颗粒。但由于缺乏有效的质量标准和监测能力,导致配方颗粒成分不清,质量参差不齐。《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》于2019年由国家药监局发布,该要求提出了“标准汤剂”这一概念,作为衡量中药免煎剂是否与临床汤剂基本一致的标准,并指出药材、饮片、中间体(浸膏或干膏粉)和免煎颗粒之间的特征性图谱应具有关联性。目前,关于红芪指纹图谱的报道多集中于基原与炮制品的研究,但红芪配方颗粒质量全程控制尚无相关研究报道。本研究建立了红芪药材、饮片、标准汤剂和配方颗粒的HPLC指纹图谱,对其相关性进行评价,并考察四者之间化学成分的差异,旨在为红芪配方颗粒替代传统饮片的可行性研究及其质量控制提供依据。
1 材料
1.1 仪器
LC-16型高效液相色谱仪(日本岛津);BT125D型十万分之一电子天平(赛多利斯科学仪器);DD-5M型低速大容量离心机(湘仪);HH-6型恒温水浴锅(西安超杰数显);SB25-12DTD型超声波清洗机(宁波新芝);YC-1000型实验室喷雾制粒包衣机(上海雅程);DZF-6090型真空干燥箱(上海一恒)。
1.2 药材
毛蕊异黄酮葡萄糖苷(批号:CHB171102)、芒柄花苷(批号:CHB171026)、毛蕊异黄酮(批号:CHB171102)、7-羟基-4'-甲氧基异黄酮(批号:CHB151026)(对照品,含量均≥98%,成都克洛玛生物科技有限公司);乙腈、甲醇、磷酸均为色谱纯(天津大茂化学试剂厂);水为超纯水;15批红芪药材均购于甘肃陇南,见表1,经甘肃中医药大学中药鉴定教研室李硕副教授鉴定为豆科植物多序岩黄芪Hedysarum polybotrys
Hand.-Mazz.的干燥根,符合2020年版《中国药典》一部相关规定。参照药典中的红芪炮制方法(除去杂质,大小分开,洗净,润透,切厚片,干燥)将15批红芪药材炮制为红芪饮片。表1 15批红芪药材来源信息
Tab 1 Source information of 15 batches of Hedysari Radix
?
2 方法与结果
2.1 红芪药材、饮片、标准汤剂与配方颗粒中有效成分的含量测定
2.1.1 色谱条件色谱柱为Wondasil C-WR(4.6 mm×150 mm,5 μm),流动相为乙腈(A)-0.1%磷酸水(B),梯度洗脱(0~7 min,15%~16%A;7~17 min,16%~18%A;17~19 min,18%~27%A;19~29 min,27%~29%A;29~31 min,29%~30%A;31~61 min,30%~60%A;61~63 min,60%~100%A),检测波长280 nm,流速1.0 mL·min,进样体积10 μL,柱温35℃。
2.1.2 混合对照品溶液的制备 精密称取毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮对照品适量,加甲醇制成每1 mL分别含0.02、0.06、0.02、0.15 mg的混合对照品溶液,即得。
2.1.3 供试品溶液的制备根据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》,制备红芪的标准汤剂(每克标准汤剂相当于2.50 g饮片),编号H1~H15,具体方法如下:取红芪饮片100 g,精密称定,加9倍量水浸泡30 min,武火煮沸,文火保持微沸30 min,趁热滤过;药渣加7倍水煎煮,武火煮沸,文火保持微沸30 min,趁热滤过,合并2次煎液,减压浓缩,冷冻干燥得标准汤剂粉未。
同法制得煎液,再减压浓缩,喷雾干燥,制备同批号的配方颗粒(每克配方颗粒相当于2.35 g饮片)。
称取15批红芪药材粗粉6.0 g、饮片粗粉6.0 g(均过4号筛),标准汤剂粉末3.0 g、配方颗粒粉末3.0 g(研细),分别置于具塞锥形瓶中,加入甲醇25 mL,密塞,称定质量,超声处理(功率500 W,频率40 kHz)60 min,放冷,补足失重,以4000 r·min离心10 min,取上清液,定容于25 mL量瓶中,即得。
2.1.4 线性关系考察 精密吸取“2.1.2”项下混合对照品溶液0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0 mL,分别置于10 mL量瓶中,加甲醇定容至刻度,摇匀,即得系列浓度混合对照品溶液。按“2.1.1”项下色谱条件测定,记录峰面积。以质量浓度(X
)为横坐标,峰面积为纵坐标(Y
)绘制标准曲线,见表2。表2 各成分线性关系
Tab 2 Linearity of constituents
成分 回归方程 r 线性范围/(mg·mL-1)毛蕊异黄酮葡萄糖苷 Y=1.18×107X-7.40×103 0.9995 0.0010~0.0080芒柄花苷 Y=1.19×107X-1.36×104 0.9996 0.0030~0.0240毛蕊异黄酮 Y=2.42×107X-9.05×103 0.9997 0.0010~0.0080 7-羟基-4'-甲氧基异黄酮 Y=3.639×107X-6.30×104 0.9996 0.0075~0.0600
2.1.5 精密度试验 取同一标准红芪药材供试品溶液(H1),连续进样6次。结果毛蕊异黄酮葡萄糖苷、毛蕊异黄酮、芒柄花苷、7-羟基-4'-甲氧基异黄酮峰面积的RSD
分别为1.3%、0.79%、1.4%、1.9%,表明仪器的精密度良好。2.1.6 重复性试验 取同一批红芪药材(H1),平行制备6份供试品溶液,进样测定。结果毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮峰面积的RSD
值分别为2.8%、1.6%、2.6%、1.4%,表明该方法重复性良好。2.1.7 稳定性试验 取同一红芪药材供试品溶液(H1),分别于0、2、4、8、12、24、48 h进样,记录峰面积。结果显示毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮峰面积的RSD
分别为1.4%、1.6%、2.4%、1.9%,表明供试品溶液在48 h内稳定。2.1.8 加样回收试验 取红芪药材(H1)约3 g,共取6份样品,精密加入适量毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮对照品,置具塞锥形瓶中,按“2.1.3”项下方法制备供试品溶液,进样测定,计算加样回收率。结果显示红芪药材中毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮平均回收率分别为99.5%、100.1%、100.8%、99.6%,RSD
分别为2.2%、1.8%、2.5%、1.3%,说明该方法回收率良好。2.1.9 样品含量测定 按“2.1.1”项下色谱条件测定15批红芪样品,结果见表3。毛蕊异黄酮葡萄糖苷(A)、芒柄花苷(B)、毛蕊异黄酮(C)、7-羟基-4'-甲氧基异黄酮(D)在15批红芪药材与饮片中的平均含量分别为13.56、25.33、9.90、185.46 μg·g与8.55、18.00、7.19、137.28 μg·g,红芪药材加工成饮片会有少量的成分损失,故红芪饮片中4种有效成分平均含量略低于药材;15批红芪标准汤剂与配方颗粒中4种有效成分的平均含量分别为51.45、93.25、18.49、159.84 μg·g与36.16、60.26、15.88、119.09 μg·g,由于加入辅料故配方颗粒4种有效成分含量偏低。
表3 含量测定结果(μg·g)
Tab 3 Content determination (μg·g)
编号 药材 饮片 标准汤剂 配方颗粒A B C D A B C D A B C D A B C D H1 14.22 25.24 10.65 178.06 9.72 23.33 9.42 171.49 43.92 84.25 15.96 133.44 31.24 71.56 17.56 123.02 H2 13.42 23.12 9.29 165.44 6.86 14.45 5.65 107.70 65.16 113.47 21.51 177.56 40.17 58.95 18.09 124.37 H3 12.85 32.88 11.61 216.59 9.62 15.42 5.76 115.55 54.58 83.13 19.40 155.33 41.29 62.91 19.53 128.77 H4 18.63 31.78 10.88 209.14 7.64 18.26 7.00 124.70 54.63 98.50 20.13 171.09 41.25 61.43 18.41 127.29 H5 12.37 22.33 9.56 192.94 7.51 14.82 5.88 117.67 66.64 109.55 21.88 187.34 36.64 53.42 14.91 117.25 H6 9.90 17.33 8.97 177.31 8.50 20.04 7.88 143.62 42.67 77.64 16.29 143.33 32.79 68.36 16.33 122.26 H7 11.24 22.72 9.93 189.66 9.67 21.45 8.89 169.40 24.75 75.49 15.47 140.80 38.06 48.43 11.80 121.70 H8 13.11 34.64 12.66 220.06 8.93 12.44 8.03 149.23 43.98 77.59 16.22 144.93 43.87 61.00 16.40 125.23 H9 11.39 17.88 6.83 141.80 8.32 18.96 7.20 144.78 41.35 73.49 15.98 145.39 37.29 67.32 14.69 113.40 H10 10.43 19.09 7.75 157.70 7.01 15.28 5.98 112.85 59.49 93.39 19.24 169.49 36.77 61.50 14.01 109.35 H11 10.28 19.21 7.57 148.00 8.61 20.56 8.56 138.45 53.05 78.51 16.77 153.16 23.68 57.21 15.48 118.63 H12 12.99 21.45 8.08 148.31 9.67 17.91 7.21 144.52 56.67 86.62 18.25 167.55 33.67 61.49 17.87 128.81 H13 17.12 29.29 10.47 198.82 8.19 18.20 7.22 137.24 61.51 103.62 18.58 165.32 37.24 64.20 15.14 114.44 H14 18.77 33.13 12.14 216.70 8.91 18.89 5.73 129.04 65.33 122.49 21.54 164.95 35.31 52.89 15.22 110.13 H15 16.67 29.87 12.11 221.34 9.15 19.92 7.47 153.02 37.96 121.01 20.10 177.89 33.14 53.25 12.73 101.65平均 13.56 25.33 9.90 185.46 8.55 18.00 7.19 137.28 51.45 93.25 18.49 159.84 36.16 60.26 15.88 119.09
2.2 红芪药材、饮片、标准汤剂与配方颗粒的HPLC指纹图谱
2.2.1 对照品溶液的制备 取“2.1.2”项下的混合对照品溶液,过0.45 μm微孔滤膜,4℃下保存备用。
2.2.2 HPLC指纹图谱的建立 15批红芪药材、饮片、标准汤剂与配方颗粒供试品溶液按“2.1.3”项下方法制备,进样测定,记录色谱图,结果见图1。

图1 15批红芪药材(A)、饮片(B)、标准汤剂(C)、配方颗粒(D)HPLC指纹图谱Fig 1 HPLC fingerprint of 15 batches of raw herbs,decoction pieces,standard decoction and dispensing granules of Hedysari Radix
2.2.3 相关性分析 将15批红芪药材、饮片、标准汤剂与配方颗粒HPLC指纹图谱数据导入“中药色谱指纹图谱相似度评价系统”(2012版),采用中位数法生成对照指纹图谱,选择时间宽度为0.1。图2为指纹图谱。标准汤剂、配方颗粒共有14个色谱峰,但均未检测到峰11、14、17、18;红芪药材、饮片共有17个色谱峰,但均未检测到峰3。经比较分析后确定红芪药材、饮片、标准汤剂与配方颗粒共有13个共有峰。通过与混合对照品紫外吸收及保留时间进行对比,确认峰5、8、12、13分别为毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮。因13号峰在红芪药材、饮片、标准汤剂与配方颗粒中分离度和峰高均最佳,故选择该峰为参照峰。
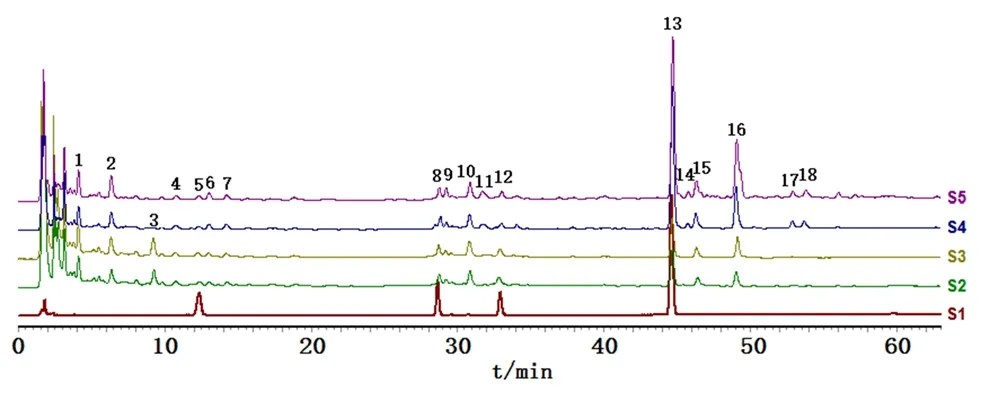
图2 混合对照品(S1)、红芪配方颗粒(S2)、标准汤剂(S3)、饮片(S4)、药材(S5)的HPLC对照指纹图谱比较Fig 2 HPLC reference fingerprint of mixed control(S1),dispensing granules(S2),standard decoction(S3),decoction pieces(S4),and raw herbs(S5)of Hedysari Radix
表4为红芪药材、饮片、标准汤剂与配方颗粒HPLC对照指纹图谱相似度结果。红芪配方颗粒与标准汤剂对照指纹图谱相似度为0.946,两者有效成分差异很小,为红芪配方颗粒质量控制提供了依据。
表4 红芪药材、饮片、标准汤剂与配方颗粒相似度结果Tab 4 Similarity of HPLC fingerprint of raw herbs,decoction pieces,standard decoction and dispensing granules of Hedysari Radix
样品 药材对照图谱饮片对照图谱标准汤剂对照图谱配方颗粒对照图谱药材对照图谱 1.000饮片对照图谱 0.988 1.000标准汤剂对照图谱 0.799 0.765 1.000配方颗粒对照图谱 0.802 0.726 0.946 1.000
2.2.4 精密度试验 取同一红芪配方颗粒供试品溶液(H1),连续进样6次,记录峰面积。以峰13为参照,计算得各共有峰相对保留时间和相对峰面积的RSD
分别小于2.5%和1.6%,表明仪器的精密度良好。2.2.5 重复性试验 取同一批红芪配方颗粒(H1),平行制备6份供试品溶液,进样测定,记录峰面积。以峰13为参照,计算得各共有峰相对保留时间和相对峰面积的RSD
分别小于1.7%和1.9%,表明该方法重复性良好。2.2.6 稳定性试验 取同一红芪配方颗粒供试品溶液(H1),分别于0、2、4、8、12、24 h进样,记录峰面积。以峰13为参照,计算得各共有峰相对保留时间和相对峰面积的RSD
分别小于1.8%和2.4%,表明供试品溶液在24 h内稳定性良好。3 讨论
3.1 色谱条件优化
本研究综合考察了乙腈-甲酸水溶液和乙腈-磷酸水溶液在不同配比条件下色谱峰的峰形和分离度情况,以乙腈-0.3%甲酸水溶液、乙腈-0.1%甲酸水溶液及乙腈-0.1%磷酸水溶液梯度洗脱时,乙腈-甲酸水溶液所得色谱峰的基线不平直,而乙腈-磷酸水溶液基线平直,出峰数量、保留时间和分离度良好;考察不同波长下(240、254、280 nm)的指纹图谱,在280 nm条件下,所得峰面积、峰形、保留时间和出峰数量最佳。
3.2 指纹图谱与特征峰
本研究共确定了13个共有特征峰,且能在5、8、12、13号峰位置与毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、7-羟基-4'-甲氧基异黄酮4种化学成分相对应。通过对红芪药材、饮片、标准汤剂与配方颗粒HPLC指纹图谱的比对发现,红芪的药材和饮片中未发现3号峰,而该成分在标准汤剂与配方颗粒中均存在,而峰11、14、17、18在标准汤剂和配方颗粒中均未检测到,但在红芪药材和饮片中均存在。这一现象可能是红芪(药材和饮片)在煎煮、浓缩和喷雾干燥过程中由于高温导致其药液中的一部分化学成分发生了转化,需要进一步研究。
3.3 红芪药材、饮片、标准汤剂和配方颗粒之间的相关性评价
配方颗粒作为中药饮片改革的新趋势,具有免煎煮、方便携带等优点。但经制剂工艺加工而成的中药配方颗粒已失去了饮片所具有的外观辨别特征,在显微结构下也不易鉴别,并且以单一成分衡量质量标准很难反映配方颗粒的整体特征。中药指纹图谱能最大限度获取配方颗粒、药材及饮片之间的化学信息,高效地将中药配方颗粒的内在信息反馈在图谱中,直观观察和评价内在质量。本试验建立红芪药材、饮片、标准汤剂和配方颗粒HPLC指纹图谱,考察其相似度,并对其相关性进行评估。结果表明,饮片与标准汤剂、配方颗粒对照图谱相似度分别为0.765、0.726,这可能与标准汤剂和配方颗粒的制备过程中要经过一系列的高温(如煎煮、浓缩干燥)处理,导致一些成分产生变化(如3号峰的出现)有关。比较发现,红芪药材与饮片的相似度为0.988,配方颗粒和标准汤剂的相似度为0.946,表明药材与饮片、配方颗粒与标准汤剂之间相似度良好,本研究可为配方颗粒替代标准汤剂提供参考。
4 小结
中药汤剂疗效显著,但存在煎煮费时、携带不便、保存困难等缺点。中药配方颗粒解决了中药汤剂的不足,作为中药饮片的一种补充形式,逐渐被人们认可。但中药配方颗粒成分复杂,质量标准难以把控。本试验将红芪配方颗粒和HPLC指纹图谱相结合,可为红芪配方颗粒的质量控制提供依据。
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国药学药品知识仓库(2022年5期)2022-04-11
农家参谋(2021年7期)2021-10-12
家庭医学(2021年12期)2021-08-23
中国药房(2021年2期)2021-02-21
食品与健康(2020年4期)2020-05-18
健康前沿(2019年6期)2019-09-10
中国食品(2018年7期)2018-09-10
家庭医药·快乐养生(2017年4期)2017-04-19
家庭百事通·健康一点通(2017年2期)2017-02-23
