
AQP2基因突变所致先天性肾性尿崩症1例并文献复习
2022-05-03 00:39陈秀清吴佩文刘东晖
中国医药科学 2022年6期
陈秀清 吴佩文 刘东晖
[摘要] 目的对 1例先天性肾性尿崩症患者进行临床及基因分析。方法对 1例自婴儿期出现多饮、多尿症状的患者进行临床和实验室检查,并采集患者及其父母、哥哥的外周血,提取基因组 DNA,用 PCR 扩增血管加压素2 受体(AVPR2)及水通道蛋白-2(AQP2)基因的全部编码区,并进行直接测序。结果患者自婴儿期开始出现脱水症状,9岁确诊为肾性尿崩症时伴发了肾功能不全、身材矮小、智力低下症状。基因检测发现患者 AQP2基因存在 c.211G>A(p.V71M)纯合子突变。结论先天性肾性尿崩症早期诊断困难,基因检测有助于早期明确诊断。
[关键词] 肾性尿崩症;水通道蛋白-2基因;基因突变;身材矮小
[中图分类号] R722.1 [文献标识码] A [文章编号] 2095-0616(2022)06-0189-04
Study on a case ofcongenital nephrogenic diabetes insipidus caused by AQP2 gene mutation and review of relevant literature
CHEN Xiuqing WU Peiwen LIU Donghui
Department of Endocrinology, the First Affiliated Hospital of Fujian Medical University, Fujian Clinical Research Center for Metabolic Diseases, Fujian Institute of Diabetes Prevention and Control, Institute of Metabolic Diseases Affiliated to Fujian Medical University, Fujian, Fuzhou 350004, China
[Abstract] Objective To analyze a patient with congenital nephrogenic diabetes insipidus (NDI) clinically and genetically. Methods A patient who presented with symptoms of polydipsia and polyuria since infancy was examined for clinical and laboratory indexes. The peripheral blood was collected from the patient, her parents and brother for genomic DNA extraction, followed by amplifying and directly sequencing all coding regions of AVPR2 and AQP2 genes with polymerase chain reaction (PCR). Results The patient had symptoms of dehydration since infancy, and was diagnosed with NDI complicated with renal insufficiency, short stature, and mental retardation at the age of 9 years old. Genetic testing revealed the presence of a homozygous nonsense mutation (c.211G>A [p.V71M]) in the AQP2 gene of the patient. Conclusion Early diagnosis of congenital NDI is difficult, and genetic testing is conducive to early and accurate diagnosis.
[Key words] Nephrogenic diabetes insipidus; AQP2 gene; Gene mutation; Short stature
先天性腎性尿崩症(congenital nephrogenic diabetes insipidus, CNDI)是一种罕见的遗传性疾病,其致病机制是由于肾脏集合管对血浆精氨酸加压素(arginine vasopressin, AVP)抵抗,从而导致尿液浓缩障碍[1]。大多数 CNDI 患者的临床表现为出生或婴儿期开始反复出现多饮、多尿、间断发热、脱水及高钠血症[2]。患者若未得到准确诊断和治疗可能会经历不可逆转的组织损伤、发育不良和智力受损[3]。
CNDI 由两种不同基因突变引起,其中约90%发生在血管加压素2 受体(vasopressin 2 receptor, AVPR2)基因,10%发生在水通道蛋白-2(aquaporin-2, AQP2)基因[2]。人类 AQP2基因位于染色体12q13区,为常染色体显性或隐性遗传。在人类 AQP2基因中已经描述了近66种致病突变[4],在我国发现了10余种 AQP2基因致病突变类型。
本研究报道1 例纯合子 AQP2基因突变(c.211G>A)的 CNDI 患者。患者从婴儿期开始反复出现脱水,但直到9 岁才被确诊,导致生长迟缓及脏器功能受损。
1 病例资料
患儿,女,9岁,以“多饮、多尿8 年余,加重1 月”为主诉于2018年 7月就诊于福建医科大学附属第一医院内分泌科。患者系第二胎第二产,足月顺产,出生体重及身长在正常范围。患者8 年余前出现多饮、多尿(具体不详),在4 岁以前还伴有不明原因的发烧、频繁呕吐、进食困难、易怒、便秘和生长迟缓,并且多次出现热性惊厥,多次因发热入住当地医院,按呼吸道感染治疗。患者4 岁时每日饮水量达6 L,每晚夜尿6 ~8 次,就诊外院多次,查尿比重为1.000~ 1.005,考虑中枢性尿崩症可能,予醋酸去氨加压素(弥凝)0. 1 m g ,qd治疗,家属自觉患儿症状稍有改善,不规律用药,后未再随访。1月前停药后,家属觉患儿症状加重,就诊福建医科大学附属第一医院,收治入院。
住院期间,患者每日饮水量7 ~10 L,夜尿8~ 10次,尿量7 ~10 L/d。入院测身高117 cm (<第 3百分位),体重31 kg(第50~ 75百分位)。患者学习成绩较差,尤其是数学。实验室检查结果如下:血钠150.1(137~ 147)mmol/L,血钾3.7 (3.50~ 5.30)mmol/L,血氯112(99~ 110)mmol/L,血钙2.65(2.11~ 2.52)mmol/L,血肌酐 35 (41~ 73)μmol/L,血白蛋白50.2(40.0~ 55.0)g/L,甲状旁腺激素6.76(1.6~ 6.9)pmol/L,促甲状腺激素4.08(0.27~ 4.2)mIU/L,游离 T3 6.71 (3.1~ 6.8)pmol/L,游离 T4 13.49(12~ 22)pmol/L,尿比重为1.002,尿蛋白阴性。
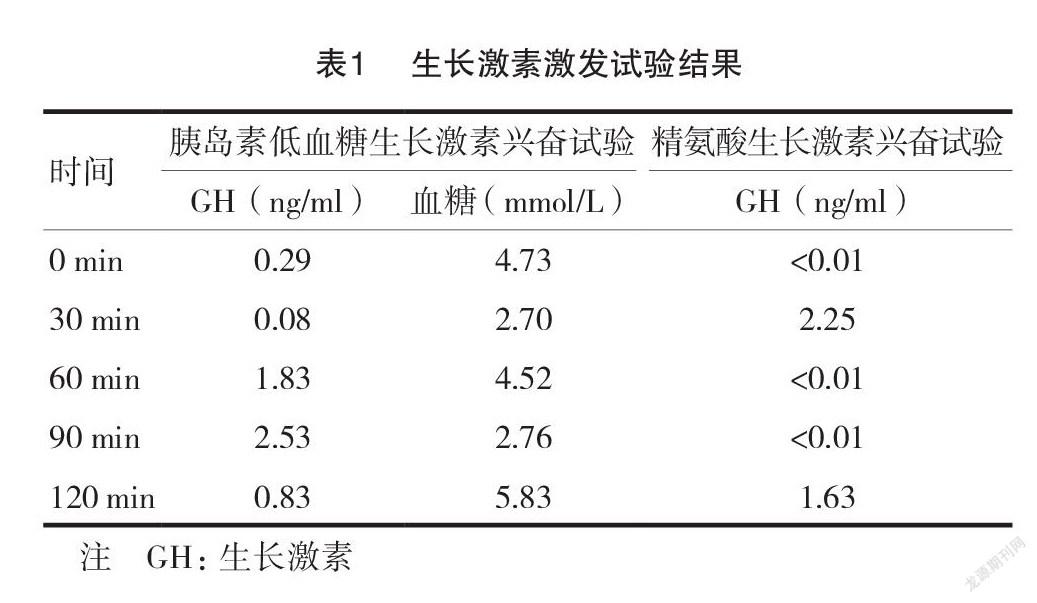
由于患者持续身材矮小,本研究对患者进行了生长激素(growth hormone, GH)激发试验,并评估血清胰岛素样生长因子-1(insulin-like growth factor-1, IGF-1)和胰岛素样生长因子结合蛋白3 (insulin-like growth factor-binding protein 3,IGFBP-3)水平。在进行胰岛素低血糖生长激素兴奋试验时,要求患者禁食12 h,次日晨8 ∶ 00静脉留置针穿刺,空腹取血后静脉注射普通胰岛素5 U,于 0、30、60、90、120 min 抽血测血糖及 GH,同时测指血血糖,结果显示 GH 峰值为2.53 ng/ml。在进行精氨酸生长激素兴奋试验时,患者禁食12 h,次日晨 8 ∶00静脉留置针穿刺,空腹取血后,将精氨酸15 g 用150 ml 注射用水稀释后对患者进行静脉滴注,30 min 输注完,于0、30、60、90、120 min 抽血测GH,结果显示 GH 峰值为2.25 ng/ml。见表1。血清 IGF-1为 201(74~ 388)ng/ml,IGFBP-3为 4.73 (1.8~ 7.1)μg/ml。
影像学检查:垂体核磁共振显示垂体高度4.6 cm,T1加权像垂体后叶高信号正常。腕部 X 线片显示患者骨龄为7 ~8 岁。肾脏超声显示双侧肾窦分离,输尿管扩张,膀胱内残留尿液达200 ml。99mTc-DTPA 动态肾显像显示左上尿路造影剂排空缓慢,左肾小球滤过率为38.8 ml/min,右肾小球滤过率为41.8 ml/min,提示肾脏排泄障碍。
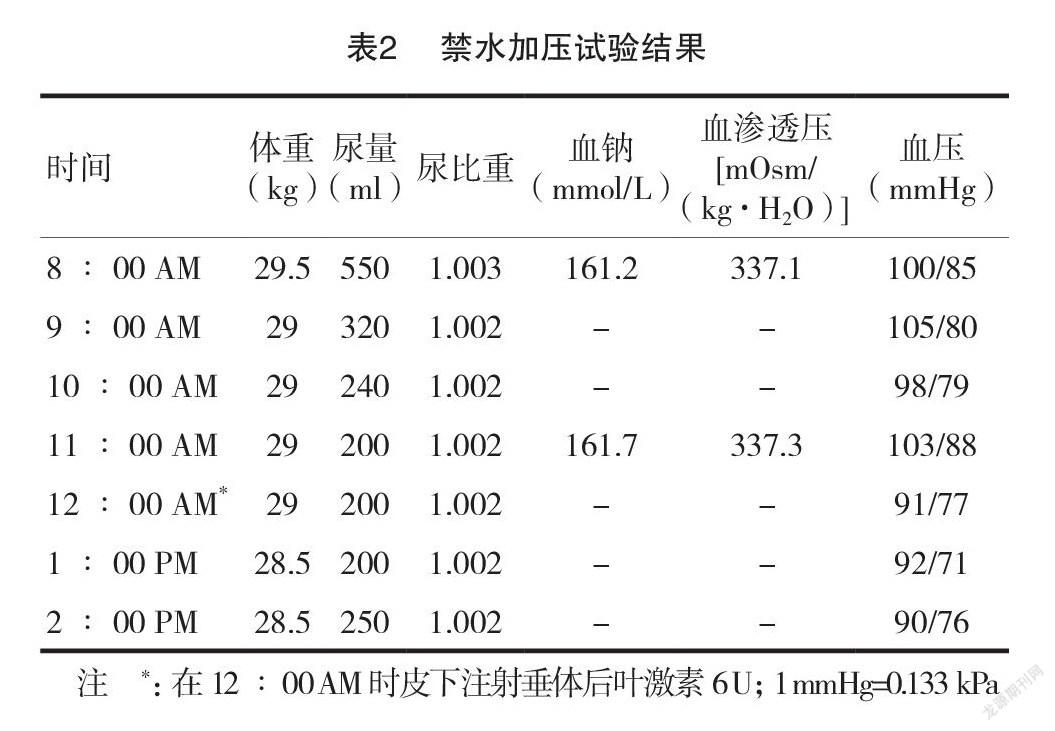
禁水加压试验如下所述。要求患者在开始试验前2 d 主动限水。试验在早上8 ∶ 00开始。前一日晚22 ∶00后不进食、不饮水。每小时监测血压、体重和尿量。测定血清渗透压和尿比重,采集尿液和血样。试验开始时,8 ∶00 AM 血钠为 161.2 mmol/L,血渗透压为337.1 mOsm/(kg·H2O);11 ∶00 AM 血鈉为161.7 mmol/L,血渗透压为 337.3 mOsm/(kg·H2O)。试验过程中,患者血钠和血浆渗透压升高,尿比重持续低平。皮下注射 6 U 垂体后叶激素之后,既没有出现尿比重增加,也没有出现尿量生成减少。禁水加压试验结果见表2 。试验结果表明患者尿崩症类型为肾性尿崩症。
经过患儿监护人知情同意,抽取患儿及其哥哥与父母外周血3 ml,应用过柱法[ 采用QIAamp blood DNA mini kit(QIAGEN 公司,美国)]提取外周血 DNA,应用 Primer Premier 5.0软件针对 AVPR2及 AQP2基因的外显子编码区设计引物,应用 2 X PCR MasterMix聚合酶(TIANGEN,天根)进行 PCR 扩增[ABI9700型 PCR 仪,Life technology(美国)],然后对 PCR 产物进行直接测序[ABI3500测序仪,Life technology(美国)],与参考序列进行比较,从而发现可能存在的基因突变。
2 基因测序结果与治疗
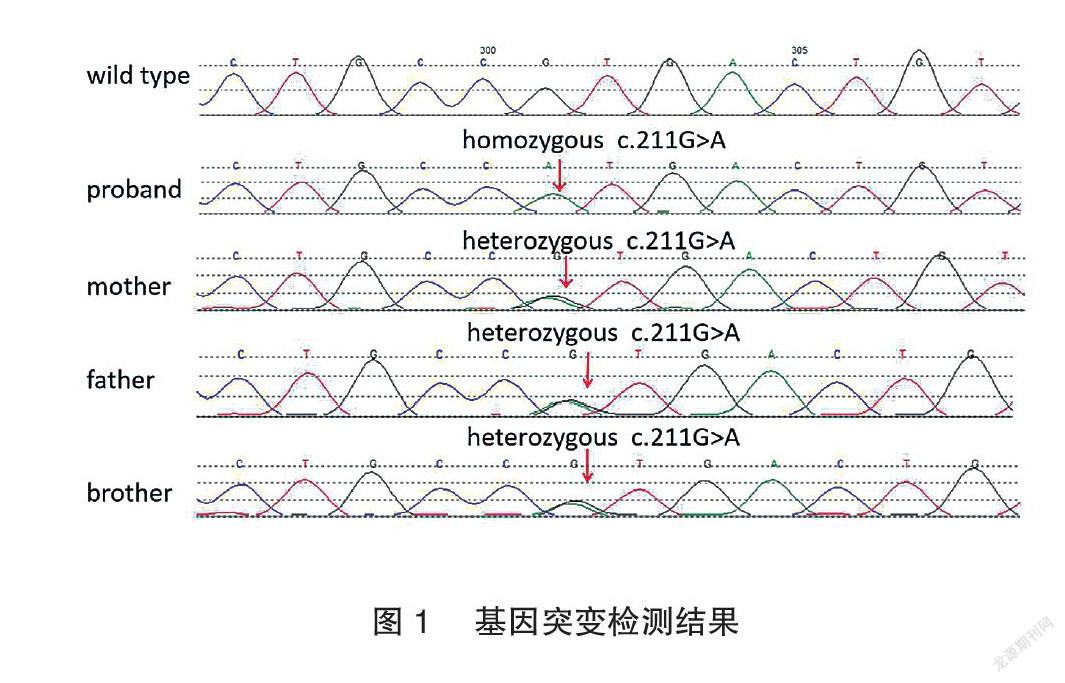
基因测序结果显示,患者 AQP2基因存在 c.211G>A(p.V71M)纯合子突变,见图1。c.211G>A 为错义突变,突变导致第71位氨基酸残基由缬氨酸变异为蛋氨酸。患者健康的父母和哥哥检测到 AQP2基因 c.211G>a(p.V71M)杂合子突变,提示该突变为常染色体隐性遗传。
患者被诊断为先天性肾性尿崩症,予口服氢氯噻嗪(上海上药信谊药厂有限公司,国药准字 H31020778,规格:25 mg)50 mg/d,联合氯化钾缓释片(上海海虹实业集团巢湖今辰有限公司,国药准字 H34021120,规格:0.5 g)1.0 g, bid 治疗,并接受低钠饮食。治疗2 个月后,患者的尿量减少到4~ 5 L/d,夜间排尿减少到2 ~3 次。治疗6 个月后,患者的尿量为4 ~5 L/d,夜间排尿2 ~3 次,身高增长 7 cm,生长速度较前加快。
3 讨论
本文报告1 例自幼多尿、多饮的患者,诊断为 CNDI,进一步的基因检测证明该患者携带 AQP2基因的 c.211G>A(p.V71M)纯合子突变。患者在婴儿期即出现反复发热、喂养困难、生长迟缓等症状,但没有得到正确的诊断和治疗。9岁确诊时,已出现器官和组织损伤,包括膀胱扩张、肾积水、肾功能不全、身材矮小。
研究顯示, CNDI 的远期并发症包括肾盂积水、输尿管积水、肾衰竭、膀胱输尿管反流、膀胱增大、神经源性膀胱、夜间遗尿等,如果病情控制不佳,反复的高渗性失水及补液过快导致的脑水肿将造成脑细胞破坏,引起智力发育障碍,若延误诊断甚至会导致生命危险[3]。鉴于先天性尿崩症的大多数病例是中枢性尿崩症,肾性尿崩症在临床上经常被误诊[5],正如本文病例。AVP 升高提示肾性尿崩症,然而, AVP 不稳定且难以测量。肾性尿崩症的核磁共振表现为垂体后叶高信号正常,在大多数中枢性尿崩症病例中,垂体后叶的高信号缺失,然而在中枢性尿崩症早期垂体后叶高信号也可能正常[6],因此,核磁共振在鉴别诊断中的价值有限。此外,大多数患者年龄太小,无法忍受禁水加压试验。因此,基因检测是加速 CNDI 准确诊断的关键。
人类 AQP2基因位于染色体12q13区,为常染色体显性或隐性遗传[7]。AQP2基因突变的类型有错义突变、移码突变、无义突变、剪接位点突变等。 AQP2基因的 c.211G>A(p.V71M)错义突变分别在一个巴基斯坦和一个突尼斯的近亲婚配家庭中被报道过[8-9]。AQP2基因编码的蛋白质由271个氨基酸组成,包含6 个跨膜结构域和1 个 COOH 末端。通常, AQP2在顶膜上形成稳定的四聚体,四聚体由四个相同的蛋白亚基组成。AQP2决定肾集合管对水的通透性并维持体内水通道平衡。AQP2 基因突变导致水渗透性功能损害,这些突变可抑制 AQP2向顶膜的穿梭,或者引起内质网(ER)的错误折叠[10]。
患者身材矮小,骨龄落后, GH 激发试验的峰值低于3 ng/ml,表明患者存在严重的生长激素缺乏症。美国哈佛大学多中心回顾性研究发现,约 70%肾性尿崩症患者的身高和体重低于正常人群的 -2SD,平均随访6 年后,仍有38%的 AVPR2 突变患者和29%的 AQP2突变患者身高和体重低于正常人群的-2SD,治疗依从性好的患儿有追赶生长[11]。本研究是国内外首次对 AQP2基因突变患者进行生长激素激发试验,发现 CNDI 存在生长激素缺乏症。北京协和医院也曾经对1 例 AVPR2基因突变的身材矮小 CNDI 患者进行低血糖生长激素检测,发现存在生长激素缺乏[12]。对 CNDI 身材矮小患者的治疗,目前主要为营养支持及纠正电解质异常,但是大多数患者不能达到预期身高[11]。
目前对 CNDI 的常规治疗包括低钠饮食、利尿剂和非甾体抗炎药等[13],但这些药物可能导致电解质失衡、胃肠道反应和肾损害等副作用,且上述治疗只是部分减少了尿量,而不是在病因水平上进行治疗。有研究提出新的靶向治疗分子可能可以治疗 CNDI,原理是试图恢复蛋白质折叠的准确性,但尚未被证明有效或达到临床应用标准[14]。基因治疗或许是未来治疗的方向[15]。
综上所述,本文报告1 例中国女孩 AQP2的 c.211G>A(p.V71M)突变引起的 CNDI。由 AQP2突变引起的 CNDI 是一种罕见的遗传病,由于大多数患者发病时年龄太小,无法耐受禁水加压试验,导致在疾病早期容易被漏诊,延迟诊断会导致严重的并发症和组织损伤,因此,对有脱水症状的儿童进行适当的基因检测是很必要的,以避免CNDI患者出现严重的并发症和不可逆的组织损伤。
[参考文献]
[1] Atmis B,Bayazit AK,Melek E,et al.From infancy toadulthood: challenges in congenital nephrogenic diabetesinsipidus[J].J Pediatr Endocrinol Metab,2020,33(8):1019-1025.
[2] Bichet DG,BockenhauerD.Genetic forms of nephrogenicdiabetes insipidus (NDI): Vasopressin receptor defect (X-linked) and aquaporin defect (autosomal recessive and dominant)[J].Best Pract Res Clin Endocrinol Metab,2016,30:263-276.
[3] Shakaroun D,Nasser H,Munie S,et al.Nephrogenicdiabetes insipidus after esophagectomy in a patient with remote history of lithium treatment: A case report[J].Int J Surg Case Rep,2019,57:71-73.
[4] Kavanagh C,UyNS.Nephrogenic Diabetes Insipidus[J].Pediatr Clin North Am,2019,66:227-234.
[5] Hu L,Yang L,Yan K,et al.Importance of EarlyGenetic Sequencing in Neonates Admitted to NICU with Recurrent Hypernatremia: Results of a Prospective Cohort Study[J].Neonatology,2022,119(1):103-110.
[6] Milano S,Carmosino M,Gerbino A,et al.HereditaryNephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update[J].Int J Mol Sci,2017,18(11):2385.
[7] Bichet DG.GENETICS IN ENDOCRINOLOGY Pathophysiology, diagnosis and treatment of familial nephrogenic diabetes insipidus[J].Eur J Endocrinol,2020,183(2):29-40.
[8] Bougacha-Elleuch N,Lassoued MB,Miled N,et al.Characterization of V71M mutation in the aquaporin-2 gene causing nephrogenic diabetes insipidus[J].J Genet,2008,87:279-282.
[9] Marr N,Bichet DG,Hoefs S,et al.Cell-biologicand functional analyses of five new Aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus[J].J Am Soc Nephrol,2002,13:2267-2277.
[10] Liao P,Xiang T,Li H,et al.Integrating PopulationVariants and Protein Structural Analysis to Improve Clinical Genetic Diagnosis and Treatment in Nephrogenic Diabetes Insipidus[J].Front Pediatr,2021,29(9):566524.
[11] D'Alessandri-Silva C,Carpenter M,Ayoob R, et al.Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study[J].Front Pediatr,2020,21(7):550.
[12] 趙丹青,平凡,朱慧娟.AVPR2基因突变所致先天性肾型尿崩伴生长激素缺乏症的临床特征[J].中华临床医师杂志(电子版),2013,7(23):10559-10562.
[13] Priya G,Kalra S,Dasgupta A,et al.DiabetesInsipidus: A Pragmatic Approach to Management[J]. Cureus,2021,13(1):e12498.
[14] Itea AM,Săsăran OM,Cozea I,et al.Nephrogenicdiabetes insipidus in children (Review)[J].Exp Ther Med,2021,22(1):746.
[15] Ando F,Uchida S.Activation of AQP2 water channelswithout vasopressin:therapeutic strategies for congenital nephrogenic diabetes insipidus[J].Clin Exp Nephrol,2018,22(3):501-507.
(收稿日期:2021-09-13)
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
科学导报(2021年29期)2021-06-03
阅读(科学探秘)(2021年3期)2021-06-02
南方周末(2021-02-04)2021-02-04
健康之家(2019年3期)2019-12-14
科海故事博览·下旬刊(2019年6期)2019-04-16
中学生理科应试(2017年6期)2017-09-27
中学生理科应试(2017年2期)2017-04-01
