
嗜铬细胞瘤/副神经节瘤核心差异表达基因的生物信息学分析鉴定
2022-05-06 11:07李雪丽曹彩霞宋洁冯文静钟丽娜杨学成
青岛大学学报(医学版) 2022年2期
关键词:基因表达
李雪丽 曹彩霞 宋洁 冯文静 钟丽娜 杨学成


[摘要]目的運用生物信息学方法分析筛选嗜铬细胞瘤/副神经节瘤(PCPG)核心差异表达基因(DEGs)。方法通过基因表达综合数据库(GEO)获取GSE60458微阵列芯片数据,应用在线NetworkAnalyst 3.0系统鉴定DEGs,KOBAS v3.0平台分析预测DEGs的作用通路,STRING v11.0工具绘制核心DEGs蛋白质相互作用网络图,GEPIA2平台验证核心DEGs表达水平。结果从GSE60458芯片中共获得1 903个DEGs,其中表达上调基因864个,表达下调基因1 039个。GO分析共富集1 593条通路,KEGG通路分析共富集145条通路。10个核心DEGs(CYP11A1、CYP11B1、CYP11B2、CYP17A1、HSD3B2、HSD3B1、STAR、SULT2A1、NR4A1、EGR4)主要参与甾类激素的生物合成过程。与Normal组相比较,PCPG组病人CYP11A1、CYP11B1、CYP11B2、CYP17A1、HSD3B2、STAR和SULT2A1等7个核心DEGs的表达水平显著下调(P<0.05),而HSD3B1、NR4A1和EGR4等3个核心DEGs表达水平无明显变化。结论本研究筛选出的核心DEGs可能影响甾类激素的生物合成,参与调控PCPG的发生发展。
[关键词]嗜铬细胞瘤;副神经节瘤;基因表达;计算生物学
[中图分类号]R736.6;R730.264[文献标志码]A[文章编号]2096-5532(2022)02-0183-06
doi:10.11712/jms.2096-5532.2022.58.063[开放科学(资源服务)标识码(OSID)]
[网络出版]https://kns.cnki.net/kcms/detail/37.1517.R.20220320.1549.004.html;2022-03-2210:11:14
BIOINFORMATICS ANALYSIS AND IDENTIFICATION OF CORE DIFFERENTIALLY EXPRESSED GENES IN PHEOCHROMOCYTOMA/PARAGANGLIOMA
LI Xueli, CAO Caixia, SONG Jie, FENG Wenjing, ZHONG Lina, YANG Xuecheng (Department of Endocrinology and Metabolism, The Affiliated Hospital of Qingdao University, Qingdao 266003, China)
[ABSTRACT]ObjectiveTo investigate the core differentially expressed genes (DEGs) in pheochromocytoma/paraganglioma (PCPG) based on bioinformatics. MethodsGene Expression Omnibus database was used to obtain GSE60458 microarray data. The online NetworkAnalyst 3.0 system was used to identify DEGs. KOBAS v3.0 was used for bioinformatics analysis to predict the pathway of DEGs, and STRING v11.0 was used to plot the protein-protein interaction network of core DEGS. The GEPIA2 platform was used to verify the expression level of core DEGs. ResultsA total of 1 903 DEGs were obtained from GSE60458 microarray, among which there were 864 upregulated genes and 1 039 downregulated genes. A total of 1 593 pathways were enriched by gene ontology analysis and 145 pathways were enriched by KEGG pathway analysis. Ten core DEGs (CYP11A1, CYP11B1, CYP11B2, CYP17A1, HSD3B2, HSD3B1, STAR, SULT2A1, NR4A1, and EGR4) were mainly involved in ste-roid hormone biosynthesis. Compared with the Normal group, the PCPG group had significant reductions in the expression levels of the 7 core DEGs of CYP11A1, CYP11B1, CYP11B2, CYP17A1, HSD3B2, STAR and SULT2A1 (P<0.05), while there were no significant changes in the expression levels of the 3 core DEGs of HSD3B1, NR4A1, and EGR4. ConclusionThe core DEGs screened out in this study may affect steroid hormone biosynthesis and participate in regulating the development and progression of PCPG.
[KEY WORDS]pheochromocytoma; paraganglioma; gene expression; computational biology
作为继发性高血压常见原因之一的嗜铬细胞瘤/副神经节瘤(PCPG)是一种少见的神经内分泌肿瘤,国内尚没有确切的患病率统计[1]。临床研究显示,有15%~17%的PCPG可以发展为转移性PCPG,而转移性PCPG治疗选择有限,预后差,通常5年存活率不到50%[2];并且肿瘤转移是导致病人死亡的主要原因[3]。世界卫生组织已将PCPG归类到有转移可能的肿瘤类目中,而术前生化检测和前期病理结果均难以判断PCPG是否具有转移性[4]。国内外学者认为,PCPG的发生发展在很大程度上是受基因组的改变驱动,如SDHx和VHL基因的改变导致三羧酸循环中断,低氧相关因子表达增加[1,5];MAX、RET和TMEM127等抑癌基因激活促进肿瘤生长基因的蛋白翻译[6-7]。PCPG具有较强的遗传背景,目前已经报道了20多个易感基因[8],约50%的病人存在胚系或体系基因突变[1]。目前常用肾上腺嗜铬细胞瘤评分(PASS)和肾上腺嗜铬细胞瘤及副神经节瘤分级系统(GAPP)评估PCPG的恶性生物学特征[9]。但在个体病人中致病基因的改变差异较大,复发或转移进展较慢,仅凭评分结果、总体生存期和无进展生存期,难以精准地对病人的疾病进展进行评估。故迫切需要从宏观角度捕捉可能与该病进展相关的基因变化,综合相关基因的内在机制判断肿瘤的发生发展,为未来开展基因信息监测、实现精准干预提供研究基础。
近年来,微阵列分析和测序技术已经成为筛选致病基因,识别具有诊断、治疗价值的生物标志物的有效技术,如LIN等[10]的研究通过微阵列芯片确定了KCNQ1和SCN2A是PCPG中的异常甲基化基因。生物信息学分析为高通量测序技术提供了快速有效的分析途径,其与液相色谱-串联质谱结合分析发现,COX4I2和PLAT蛋白与PCPG供血高度相关[11]。NCBI基因表达综合数据库(GEO)可以提供大量针对各种人类肿瘤的微阵列分析数据和下一代测序数据,储存了大量未被挖掘的差异表达基因(DEGs)。探究与PCPG相关的特异性DEGs及其可能参与的病理功能有助于更好地理解PCPG的进展。本研究基于GEO数据库的微阵列芯片数据,通过生物信息学分析筛选核心DEGs并探讨其可能的分子机制,以期为PCPG的发生发展提供新的研究视角。
1资料与方法
1.1微阵列芯片初始数据的获取
通过GEO数据库获取PCPG基因微阵列芯片(GSE60458)数据,其中共包含62 976个探针的初始数据,来自于12例PCPG组织样本(PCPG组,良性9例,恶性3例)和3例正常肾上腺髓质组织样本(Normal组)。并且通过GPL13607平台(Agilent-028004 SurePrint G3 Human GE 8x60K Microarray)识别探针信号对应的基因名称。
1.2DEGs的筛选
利用在线NetworkAnalyst 3.0系统,对微阵列芯片初始数据进行标准化和背景校正处理。使用该系统的Limma包对PCPG组和Normal组做层次聚类分析,以log2|Fold Change|>2且P<0.05的标准筛选DEGs,以热图显示DEGs表达谱,以火山图展示所有DEGs的显著性差异。并以DEGs为变量进行主成分分析(PCA),观察PCPG组和Normal组样本的差异。
1.3DEGs功能预测
为探究DEGs可能参与的通路,将全部DEGs输入KOBAS v3.0(KEGG Orthology Based Annotation System v3.0)线上数据平台,进行在线GO(Gene Ontology)、KEGG(Kyoto Encyclopedia of Gene and Genomes)通路富集分析,以P<0.05和错误发现率(FDR)<0.05为基因富集通路有统计学意义。
1.4核心DEGs筛选
使用STRING v11.0工具将表达差异最大的40个DEGs进行蛋白质相互作用(PPI)网络分析,将连接于模块中的基因作为核心DEGs,并将PPI网络可视化。
1.5核心DEGs表达水平验证
为验证GSE60458微阵列芯片结果的准确性,在GEPIA2平台上TCGA和GTEx数据库中获取182例PCPG标本(Tumor组)和3例正常對照标本(Normal组),分析两组核心DEGs的表达情况。使用Origin 2016软件构建生物信息学分析结果图,采用双侧t检验统计组间的差异,以P<0.05为差异具有统计学意义。
2结果
2.1PCPG差异基因表达及分布特征
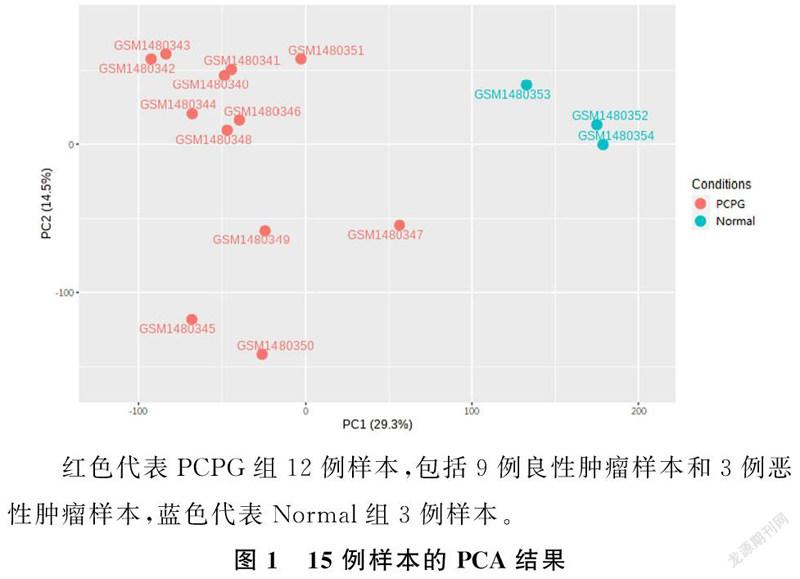
PCA显示,DEGs将两组样本从两个主成分维度(PC1、PC2)完全分离,说明DEGs表达模式具有特异性,可用于完全区分PCPG组织和正常肾上腺组织。同时观察到PCPG组9例良性肿瘤样本和3例恶性肿瘤样本未能完全分离,说明良恶性肿瘤组织DEGs表达模式的特异性差。见图1。
分析GSE60458芯片中的所有基因的表达结果,DEGs共有1 903个(图2),其中表达上调基因864个,下调基因1 039个(图3)。表达上调DEGs前10位依次为EGR、SCRT2、C1QL1、SOHLH1、CD163L、lincRNA:chr5:1731、CHRNA4、TLX3、NR4A1和ENST0000031680,表达下调DEGs前10位依次为HSD3B1、HSD3B2、MRAP、AADAC、CYP11B2、GSTA5、LOC391081、CYP11B1、MGST1和KCNK2(图4)。
2.2DEGs的GO和KEGG富集分析
GO富集分析结果显示,DEGs富集于1 593条通路,DEGs富集度最大的5条通路分别为蛋白结合(Protein binding)、细胞质膜(Plasma membrane)、胞外区(Extracellular region)、细胞膜的有机构成(Integral component of membrane)和细胞外间隙(Extracellular space)。见图5。KEGG通路富集于145条通路,其中富集度最大的5条通路分别为代谢途径(Metabolic pathways)、细胞因子-细胞因子受体相互作用(Cytokine-cytokine receptor interaction)、病毒蛋白与细胞因子及细胞因子受体的相互作用(Viral protein interaction with cytokine and cytokine receptor)、癌症通路(Pathways in cancer)和药物代谢-细胞色素P450(Drug metabolism-cytochrome P450)。见图6。
2.3核心DEGs筛选
本研究采用STRING v11.0工具软件对表达差异最大的40个DEGs进行PPI网络分析,并将PPI网络可视化。PPI网络分析结果显示,CYP11A1、CYP11B1、CYP11B2、CYP17A1、HSD3B2、EGR4、STAR、SULT2A1、NR4A1和HSD3B1等10个基因位于紧密连接的模块中,认定为核心DEGs(图7)。该模块由10个节点和32条连接线组成,结合GO和KEGG富集分析结果,CYP11B1、CYP11B2、CYP11A1、CYP17A1、HSD3B1和HSD3B2基因参与甾类激素生物合成(Steroid hormone biosynthesis),CYP11B1、CYP11B2基因参与氧化还原过程(Oxidation-reduction process),CYP11B1、CYP11B2和HSD3B2基因共同参与钙离子结合(Calcium ion binding),CYP11B1、HSD3B2基因以及HSD3B1基因参与代谢通路(Metabolic pathways),NR4A1基因参与MAPK信号通路(MAPK signaling pathway)。
2.4核心DEGs表达水平验证
利用TCGA和GTEx数据库,对GSE60458芯片的分析结果进行验证。与Normal组相比,Tumor组病人CYP11A1、CYP11B1、CYP11B2、CYP17A1、HSD3B2、STAR和SULT2A1等7个核心DEGs的表达水平显著下调(P<0.05),与GSE60458数据集的结果相同;而HSD3B1、NR4A1和EGR4等3个核心DEGs表达水平无明显变化,与GSE60458数据集的结果相似。见图8。
探究肿瘤组织和正常组织之间的差异是肿瘤研究的主要内容。PCPG是少见的神经内分泌肿瘤,由于临床数据及组织标本难以大量获得,这限制了科研人员对该病的探究。肿瘤基因微阵列芯片中包含了大量有潜在研究价值的信息,生物信息学的运用可以从宏观角度对有潜在研究价值的基因进行筛选,进而加快研究进程。但随着高通量技术的普及和大数据库的发展,“数据泛滥”成为生物信息学分析的一个难点。而PCA可以将多维度数据有效地降维分析[12]。本研究对所有样本进行了PCA,观察到PCPG组和Normal组可以从两个主成分维度完全分离,说明PCPG组织和正常肾上腺组织在基因表达模式上具有显著的差异。而PCPG组9例良性肿瘤样本和3例恶性肿瘤样本则未能完全分离,提示PCPG良恶性组织基因表达模式差异较小。日本学者统计发现,有60%的恶性PCPG最初被诊断为良性PCPG[4],本研究PCA结果也从侧面验证了这一事实。
本研究从GSE60458芯片数据集中共确定了1 903个DEGs,表明肿瘤的形成过程是一个涉及癌基因、抑癌基因和其他基因改变的复杂过程。DEGs通路富集分析进一步表明,DEGs主要参与了与肿瘤相关的生物学过程,如胞外外泌体、炎症反应、细胞因子-细胞因子受体相互作用、趋化因子信号、Ras信号通路以及MAPK信号通路等。有研究发现,PCPG病人的循环外泌体中存在双链DNA片段,且外泌体DNA片段与肿瘤细胞易感基因具有相同的突变[13];NRF2基因激活可以通过抑制氧化应激和促进肿瘤炎症来降低癌症风险[14];白细胞介素-33(IL-33)通过调节趋化因子,招募、激活固有免疫细胞,从而创造促肿瘤环境[15];在肺癌研究中,Treg细胞中IC(Immune checkpoint)分子表达水平升高,程序性死亡受体1(PD-1)表达增加,表现出强大的抑制活性[16]。近年来,PCPG病因相关基因的发现使转移性PCPG的诊断和治疗取得了重大进展,为了精准地对疾病进展进行个体化基因信息监测,还需要更为高效的分析技术。
由于筛选出的1 903個DEGs的信息众多,为精确研究范围,本研究通过STRING v11.0工具将表达差异最大的40个DEGs进行PPI网络分析,结果显示,10个核心DEGs存在着直接或间接地相互
作用。表明核心基因表达水平的变化有可能干扰PCPG的发生和发展。通过TCGA和GTEx数据库验证10个核心基因表达水平,结果与GSE60458芯片结果一致。其中HSD3B1、NR4A1和EGR4基因的表达水平两组间差异无统计学意义,可能是由于TCGA和GTEx数据库中正常对照组纳入的样本数量有限,数据存在偏移,提示仍需收集肿瘤标本和临床数据,进一步验证分析结果的准确性。
本文富集分析结果显示,CYP11B1、CYP11B2、CYP11A1、CYP17A1、HSD3B1、HSD3B2等基因共同参与甾类激素的生物合成。类固醇生成酶分为细胞色素P450和羟基类固醇脱氢酶/酮类固醇还原酶两类,包括CYP11A1、CYP11B1、CYP11B2、HSD3B2、HSD11B1、HSD11B2等[17-18]。甾类激素生物合成是在多种酶和辅助因子的参与下,由胆固醇经多步酶促反应转化为具有生物活性的甾类激素,涉及多种细胞的生理功能。胆固醇是免疫细胞膜的基本脂质成分,可以通过T细胞获得,并参与细胞激活、增殖、代谢等[19]。已有研究发现,肿瘤胆固醇通过调节内质网应激通路[20],或诱导CD8+T细胞功能衰竭[21],使抗肿瘤免疫失活;氧甾醇通过与核肝X受体(LXR)α和LXRβ相互作用,发挥内源性脂质代谢调节作用,招募促血管生成和免疫抑制的中性粒细胞[22],建立促肿瘤微环境;腺体外类固醇激素合成失调降低了免疫细胞自身的抗肿瘤能力[23]。肿瘤细胞可通过破坏免疫细胞功能,造成抗肿瘤免疫反应钝化,从而逃避免疫杀伤。此外,转录因子也可能参与抗肿瘤免疫。NR4A1基因位于12号染色体,有12个外显子,编码核转录因子。有文献报道,NR4A1转录因子在耐受性T细胞中表达增加,缺失NR4A1则可降低T细胞的耐受性,增强对肿瘤的免疫力[24]。本研究芯片分析结果显示,NR4A1表达上调,通路富集显示NR4A1参与MAPK信号通路,可能通过影响PCPG激酶信号通路[1],参与肿瘤的发生。
本研究分析報告了两个在PCPG中鲜有研究的DEGs。SOHLH1基因位于9号染色体,其编码蛋白具有转录因子的功能。LIU等[25]研究发现,Sohlh1蛋白在胶质母细胞瘤中表达下调,且与胶质母细胞瘤的恶性程度呈负相关。体外实验进一步确定了Sohlh1通过抑制Wnt/β-catenin信号通路抑制胶质母细胞瘤细胞的增殖、迁移和侵袭[25]。Wnt信号通路在PCPG中已有深入的研究,如CSDE1体细胞突变及MAML3的体细胞基因融合激活Wnt和Hedgehog信号通路[3],使PCPG表现为强侵袭性。SOHLH1基因表达上调,可能参与PCPG的转移,提示该基因可能成为肿瘤行为的预测因子。SCRT2基因编码Scratch家族转录抑制因子2,位于20号染色体。研究发现,Scratch1和Scratch2由黏附分子E-cadherin介导,通过上皮-间充质转化机制,参与神经上皮细胞向神经元迁移或向中间神经元祖细胞转化[26]。但SCRT2在PCPG中的作用机制还有待进一步探究。
综上所述,本研究筛选出的10个核心DEGs可能影响甾类激素的生物合成,参与调控PCPG的发生发展,这为预测PCPG的潜在生物标志物提供了研究依据。
[参考文献][1]王卫庆,宁光,吕朝晖,等. 嗜铬细胞瘤和副神经节瘤诊断治疗专家共识(2020版)[J]. 中华内分泌代谢杂志, 2020,36(9):737-750.
[2]CORSSMIT E P M, SNEL M, KAPITEIJN E. Malignant pheochromocytoma and paraganglioma: management options[J]. Current Opinion in Oncology, 2020,32(1):20-26.
[3]CRONA J, TA?EB D, PACAK K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification[J]. Endocrine Reviews, 2017,38(6):489-515.
[4]TANABE A, NARUSE M. Recent advances in the management of pheochromocytoma and paraganglioma[J]. Hypertension Research, 2020,43(11):1141-1151.
[5]DWIGHT T, NA U, KIM E, et al. Analysis of SDHAF3 in familial and sporadic pheochromocytoma and paraganglioma[J]. BMC Cancer, 2017,17(1):1-12.
[6]FISHBEIN L, LESHCHINER I, WALTER V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma[J]. Cancer Cell, 2017,31(2):181-193.
[7]JOCHMANOVA I, PACAK K. Genomic landscape of pheochromocytoma and paraganglioma[J]. Trends in Cancer, 2018,4(1):6-9.
[8]BUFFET A, BURNICHON N, FAVIER J, et al. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma[J]. Best Practice & Research Clinical Endocrinology & Metabolism, 2020,34(2):101416.
[9]NEUMANN H P H, YOUNG W F, ENG C. Pheochromocytoma and paraganglioma[J]. The New England Journal of Medicine, 2019,381(6):552-565.
[10]LIN D Q, LIN J L, LI X X, et al. The identification of diffe-rentially expressed genes showing aberrant methylation patterns in pheochromocytoma by integrated bioinformatics analysis[J]. Frontiers in Genetics, 2019,10:1181.
[11]SUN F K, ZHUO R, MA W M, et al. From clinic to mechanism: proteomics-based assessment of angiogenesis in adrenal pheochromocytoma[J]. Journal of Cellular Physiology, 2019,234(12):22057-22070.
[12]GIULIANI A. The application of principal component analysis to drug discovery and biomedical data[J]. Drug Discovery Today, 2017,22(7):1069-1076.
[13]WANG L, LI Y, GUAN X, et al. Exosomal double-stranded DNA as a biomarker for the diagnosis and preoperative assessment of pheochromocytoma and paraganglioma[J]. Molecular Cancer, 2018,17(1):128.
[14]HE F, ANTONUCCI L, KARIN M. NRF2 as a regulator of cell metabolism and inflammation in cancer[J]. Carcinogenesis, 2020,41(4):405-416.
[15]DE BOECK A, AHN B Y, DMELLO C, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression[J]. Nature Communications, 2020,11:4997.
[16]KIM H R, PARK H J, SON J, et al. Tumor microenvironment dictates regulatory T cell phenotype: upregulated immune checkpoints reinforce suppressive function[J]. Journal for Immunotherapy of Cancer, 2019,7(1):339.
[17]YANCU D, SANDERSON T. Essential oils disrupt steroidogenesis in a feto-placental co-culture model[J]. Reproductive Toxicology (Elmsford, N Y), 2019,90:33-43.
[18]BACILA I A, ELDER C, KRONE N. Update on adrenal ste-roid hormone biosynthesis and clinical implications[J]. Archives of Disease in Childhood, 2019,104(12):1223-1228.
[19]BIETZ A, ZHU H Y, XUE M M, et al. Cholesterol metabolism in T cells[J]. Frontiers in Immunology, 2017,8:1664.
[20]PICARDA E, REN X X, ZANG X X. Tumor cholesterol up, T cells down[J]. Cell Metabolism, 2019,30(1):12-13.
[21]MA X Z, BI E G, LU Y, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment[J]. Cell Metabolism, 2019,30(1):143-156.e5.
[22]MORESCO M A, RACCOSTA L, CORNA G, et al. Enzymatic inactivation of oxysterols in breast tumor cells constraints metastasis formation by reprogramming the metastatic lung microenvironment[J]. Frontiers in Immunology, 2018,9:2251.
[23]MAHATA B, PRAMANIK J, VAN DER WEYDEN L, et al. Tumors induce de novo steroid biosynthesis in T cells to evade immunity[J]. Nature Communications, 2020,11:3588.
[24]LIU X D, WANG Y, LU H P, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction[J]. Nature, 2019,567(7749):525-529.
[25]LIU X Y, GAO Q, ZHAO N, et al. Sohlh1 suppresses glioblastoma cell proliferation, migration, and invasion by inhibition of Wnt/β-catenin signaling[J]. Molecular Carcinogenesis, 2018,57(4):494-502.
[26]ITOH Y, MORIYAMA Y, HASEGAWA T, et al. Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition-like mechanism[J]. Nature Neuroscience, 2013,16(4):416-425.
(本文編辑马伟平)
猜你喜欢
江苏农业科学(2017年1期)2017-02-27
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
中国实用医药(2016年30期)2016-12-28
科技视界(2016年16期)2016-06-29
江苏农业科学(2015年5期)2015-10-20
