
去氢枞酸菲环结构改性及衍生物活性研究进展*
2022-05-17 07:16韦有杰莫秀庆
云南化工 2022年4期
韦有杰,马 璐,莫秀庆,雷 禄
(百色学院 预科教育学院,广西 百色 533000 )
松香是我国具有优势可再生的林化资源,经歧化改性后可得到歧化松香,去氢枞酸是歧化松香的主要成分[1-2]。去氢枞酸含有三环菲骨架结构,本身具有良好的生物活性,因而成为天然产物改性研究的热点化合物。据报道,去氢枞酸及其衍生物具有抗菌、抗癌、消炎、抗衰老、抗溃疡等[3-10]多种生物活性。其结构中的羧基,因易于酰氯化、还原胺化、酯化等反应,而成为去氢枞酸改性的热点活性中心。通过对羧基进行改性,可以引入噁唑、噻唑、三唑、咔唑、咪唑、喹啉、吲哚和呋喃等基团,得到去氢枞酸基系列杂环衍生物。随着研究的深入,人们逐渐对去氢枞酸菲环结构的改性产生了浓厚兴趣,其中以B环和C环为重点。改性途径主要是对B环C7用CrO3氧化引入羰基[11-12],或对C环的C12和C14进行傅克酰基化[13]、卤化[14-15]、硝基化[15-16]、磺化[17],对C13上异丙基进行脱去[18]等,得到的目标化合物也表现出良好的生物活性或光学活性。本文对2015年以来在菲环结构改性上取得的研究成果进行简要总结,为去氢枞酸菲环结构改性的深入研究提供参考。去氢枞酸的结构见图1。

图1 去氢枞酸的结构
1 基于B环的改性及其衍生物活性
目前,基于去氢枞酸B环的改性主要集中于C7上,通过CrO3将之氧化成羰基,再与硫脲、氨基脲、肼、苯肼、羟胺等反应,进一步制得所需要的目标化合物。
Luo 等[19]以去氢枞酸为原料,用CrO3将B环上C7氧化得到羰基,再与苯肼反应,最后得到一种新型N取代1H-二苯并[a,c]咔唑衍生物1(图2)。该课题组采用CCK-8法和流式细胞术检测了衍生物1对胃癌细胞的杀伤作用,发现其对胃癌细胞具有很强的细胞毒性,质量浓度为 10 μg/mL 时完全抑制达 48 h 甚至更久。研究认为其作用机理是通过剂量和时间依赖性抑制细胞增殖,破坏细胞膜的完整性,并激活钙蛋白酶-1抗体自溶,以达到诱导凋亡蛋白分裂。Chen等[12]也合成了一系列含不同N-(哌嗪-1-基)烷基侧链的去氢松香酸基1H-二苯[a,c]并咔唑衍生物,并研究了衍生物对3种人肝癌细胞系(SMMC-7721、HepG2和Hep3B)的体外抗癌活性。其中,化合物2(图2)抗癌活性最强,IC50值分别为(1.39±0.13)、(0.51±0.09)mmol/L和0.73±0.08 mmol/L。还发现该化合物可提高细胞内ROS水平,降低线粒体膜电位,破坏细胞膜完整性,并最终导致HepG2细胞的癌性和凋亡。

图2 Luo等和Chen等合成的衍生物
Huang等[20]对B环C7进行氧化后与羟胺反应,并最终合成得到一系列新型具有抗肿瘤活性的去氢枞酸基手性二肽衍生物3(图3)。并采用MTT法测定化合物对人肺癌NCIeH460、宫颈癌上皮细胞HeLa和胃癌MGC-803细胞的体外抑制活性。抗肿瘤活性测试表明,多个化合物对这三种肿瘤细胞株均表现出中高水平的抑制活性。大部分化合物的抑肿瘤活性高于商业抗癌药物5-氟尿嘧啶(5-FU)。Vahermo 等[21]先将去氢枞酸C7氧化改性成羰基,再与羟胺反应,得到一系列具有不同侧链氨基酸骨架的去氢松香酸衍生物4(图3)。衍生物对多氏利什曼原虫和克氏锥虫具有较强的抗原虫活性。其中,4a和4b杀死细胞内多氏利什曼原虫IC50值分别为2.3、6.6 μmol/L。4a、4b和4c杀死细胞内克氏锥虫IC50值分别为4.2、3.9、5.8 μmol/L。

图3 Huang等和Vahermo等合成的衍生物
2016年,陈乃源等[7]以去氢枞酸为原料,经对B环C7氧化成羰基后,再对羰基α碳进行溴化并与硫脲等反应,最后合成得到一系列新型去氢枞酸基B环并噻唑-酰胺化合物。并研究了化合物对黄瓜枯萎、番茄早疫、苹果轮纹、花生褐斑及小麦赤霉等5种病原菌的抑制活性。结果表明,化合物5(图4)及其中间体(去氢枞酸基环并噻唑-胺)在50 mg/L下对苹果轮纹病菌的抑制活性均达A级,抑制率分别为90.0%和92.4%。2020年,陈乃源等[11]又在5的基础上合成得到一系列新型(芳基)甲基胺脱氢枞酸基B环融合噻唑衍生物6(图4),以CCK-8法研究了它们对HepG2、SCC9和293T的体外细胞毒活性。其中,化合物6a和6b对癌细胞具有一定的抑制活性,而对正常细胞毒性较弱,同时对PI3K/AKT/mTOR信号通路有抑制活性。
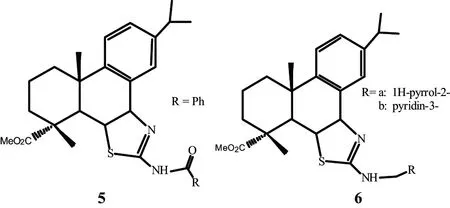
图4 文献[7]和[11]合成的衍生物
2 基于C环的改性及其衍生物活性
C环为苯环。对C环的改性方法主要是利用亲电试剂与苯环进行如卤化、硝化、磺化、傅克酰基化和脱异丙基等亲电取代反应,最后合成得到苯并杂环、氨基取代物等具有生物活性的衍生物。
Gu 等[15]对去氢枞酸C环进行卤化、脱异丙基化、硝化等改性后,合成得到化合物7(图5),并通过X射线单晶衍射研究了化合物的晶体结构,证实该化合物为单斜晶系。初步抗肿瘤实验表明,该化合物对HepG2和SMMC-7721细胞具有较强的抑制活性,IC50值分别为17.1、10.2 μmol/L。

图5 文献[16]、[17]合成的衍生物
Huang 等[17]对C环进行磺化后,合成得到一系列含有磺胺基的去氢松香酸二肽衍生物8(图5)。抗肿瘤活性研究表明,部分化合物具有较好的抑制活性,甚至优于商品化抗癌药物5-FU。其中,8a对HepG2细胞的抑癌活性最好,IC50为(4.18±1.08) μmol/L。研究发现,8a是通过抑制MMP-3活性和诱导细胞凋亡,对HepG2细胞系产生了最强的抑制作用。
Wei等[16]在去氢枞酸C14硝化还原胺化后,并最终制备一系列新颖的C-14 1,2,3-三唑去氢枞酸衍生物9(图6)。体外抗增殖活性评价表明,大多数化合物对癌细胞均有较强的抑制活性。其中,9a、9b和9c在低浓度下对耐阿霉素MCF-7生物同样有效,且呈剂量相关。特别是,结构中具有3-(叔丁基羰基氨基)苯基取代的三唑基团的最有效化合物9c,不仅对癌细胞抑制效果明显,IC50值在0.7~1.2 mmol/L 之间,且对正常细胞表现出非常弱的细胞毒性。
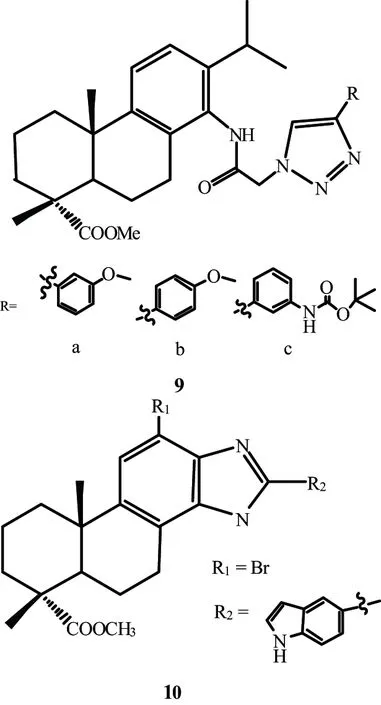
图6 文献[16]、[22]合成的衍生物
Miao 等[22]对去氢枞酸C12进行卤化,脱去C13上异丙基并对C13,C14进行硝化后,最终合成得到多个新型2-芳基苯并咪唑衍生物。采用MTT法测定了目标化合物对肝癌细胞株SMMC-7721、MDA-MB-231、HeLa、CT-26和正常肝细胞QSG-7701的体外细胞毒活性。其中,化合物10(图6)的细胞毒活性最强,其IC50值分别为0.08±0.01、0.19±0.04、 0.23±0.05、0.42±0.07 μmol/L,且对人正常肝细胞QSG-7701的抗肿瘤活性相对安全。微管蛋白聚合抑制实验和免疫荧光染色进一步证实化合物10可明显抑制微管聚合,从而破坏癌细胞的微管网络。研究还发现,该化合物10可诱导SMMC-7721细胞凋亡。
Pilar 等[18]报道,以去氢枞酸为原料,首次合成了cassane型二萜taepeenin F11(图7)。并对合成反应关键步骤甲酰基取代脱氢枞烷衍生的异丙基进行了补充研究,提出了该反应的假定机理。
檀贯妮等[23]以去氢枞酸为起始物,能过酯化、溴化、硝化、还原和C-N 偶联反应合成得到 13-[N, N-(4-萘基苯基)-苯基]胺基-脱异丙基脱氢枞酸甲酯12a及13-[N, N-双(4-萘基苯基)-苯基]胺基-脱异丙基脱氢枞酸甲酯12b两个化合物(图7)。并利用Gaussina 09程序采用密度泛函DFT/B3LYP等方法,研究了化合物结构与光谱性能之间的关系。通过对比发现,去氢枞酸骨架和萘环的引入对化合物平面性有影响,而萘环则会增大化合物的共轭程度。在甲醇、二氧六环等5种溶剂中的荧光及紫外光谱测试表明,化合物在不同极性的溶剂中荧光及紫外均变化较大,表明它们均存在溶致变色效应。该课题组[24]又以同样的方法合成得到一种含双萘的脱氢枞酸三芳胺化合物13-[N,N-双(α-萘)]胺基-脱异丙基脱氢枞酸甲酯13(图7),荧光及紫外光谱测试同样表明其具有溶致变色效应。
Yang等[25]合成系列2-芳基硫代和2-芳基胺- 1H -苯并[d]咪唑去氢枞酸衍生物,其中2-芳基胺- 1H -苯并[d]咪唑去氢枞酸衍生物14(图7)对四种癌细胞(HCT-116、MCF-7、HeLa和HepG2)抑制活性最强,IC50值分别为0.18±0.03、0.43±0.05、0.71±0.08、0.63±0.09 μmol/L,对人胃黏膜细胞系gs -1的细胞毒性显著降低,IC50为 21.95±0.73 μmol/L。此外,该化合物还可增加细胞内活性氧水平,降低线粒体膜电位,上调Bax和cleaved caspase-3 - 9水平,下调Bcl-2水平,并以剂量依赖性的方式诱导HCT-116细胞凋亡。

图7 文献[18]、[23]、[24]、[25]合成的衍生物
3 同时对B环和C环的改性及其衍生物活性
Damir等[4]首次报道了从去氢枞酸甲酯出发,在C14和C7分别引入羟基和羰基,半合成得到一种二萜类化合物(+)-流相-萜酸15(图8)及若干个类似物。根据不同的官能团对以上化合物进行了生物学比较,得出了一些基本的构效关系(SAR)。通过对6种具有代表性的人类肿瘤实体细胞(A549、HBL-100、HeLa、SW1573、T-47D和WiDr)、5种白血病细胞模型(nom -06、KOPN-8、SUP-B15、UoCB1和BCR-ABL)和4种利什曼原虫(L. infantum、L. donovani、L. amazonensis和L. guyanensis)的生物学特性进行研究,表明这些枞烷二萜类化合物可从廉价的手性化合物库中获得具有良好药理特性的新型生物活性分子。

图8 文献[4]、[26]、[27]、[14]合成的衍生物
Liu 等[26]以在去氢枞酸B环中引入肟结构和C环引入芳酰基得到16(DHAD)(图8),并与ZnAlTi-LDH组装得到DHAD/ZnAlTi -LDH复合材料。研究表明,该复合材料具有显著的活性氧(ROS)生产能力,在可见光照射下均能杀灭革兰氏阴性菌和阳性菌。同时,能有效保护皮肤免受紫外线的伤害。
Zhang 等[27]以去氢枞酸为原料在B环C7引入肟和在C14进行溴化,最后合成了一系列脱氢枞酸的7- N -酰氨乙基/丙肟衍生物,并对其对金黄色葡萄球菌纽曼株和多药耐药株(NRS-1、NRS -70、NRS-100、NRS-108和NRS-271)的抑菌活性进行了研究。大多数含三氟甲基苯基/苄基、卤素取代噻吩基、苯并噻吩基和吡咯基的目标化合物都表现出较强的体外抗菌活性。其中,化合物17a、17b和17c(图8)对5种耐多药金黄色葡萄球菌具有较高的抑菌活性,最低抑菌质量浓度(MIC)为1.25~3.13 mg/mL。Zhang 等[14]于2017年合成得到一系列去氢枞酸的N-磺胺乙基肟衍生物,并研究了化合物对金黄葡萄球菌纽曼株和耐多药菌株的抗菌活性。大多数含氯、溴、三氟甲基苯基的目标化合物具有较强的体外抗葡萄球菌活性。其中,化合物18(图8)对金黄葡萄球菌纽曼株表现出最高的抑菌活性,MIC为0.39~0.78 μg/mL。对5种耐多药金黄色葡萄球菌的抑菌活性在0.78~1.56 μg/mL 之间。
Cui等[28]以去氢枞酸为原料一系列脱氢枞酸的N -酰基氨基烷基肟衍生物19(图9),并在CHO-K1细胞的实验系统中评估了对BK通道的开启活性。构效研究表明,2-噻吩的S原子与羰基O原子之间的非共价相互作用可能对其与离子通道的相互作用造成限制。

图9 文献[28]合成的衍生物
4 展望
去氢枞酸本身具有多种生物活性,且又是天然产物,具备较好的改性研究先天条件。根据当前对菲环结构的改性取得的研究成果,对菲环进行改性是寻找具有优良生物活性的较好选择。菲环改性有B环及C环两个区域可供选择,也可同时进行改性,具有较为广阔的研究前景。因C环是苯环可进行卤化、酰基化、烷基化、硝化、磺化等反应,取得的成果相较B环要多,活性类型不只有生物活性还有光学活性。可以预见,对C环的改性的研究仍会较B环多,找到具有优良生物活性的先导物也不是不可能。引入的杂环多见含氮杂环,或多见单个杂原子的杂环,具有优良生物活性的含硫,含氧多杂原子杂环引入还比较少,这方面的研究具有较大前景。
猜你喜欢
建材发展导向(2021年15期)2021-11-05
食品安全导刊(2021年21期)2021-08-30
能源工程(2021年1期)2021-04-13
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
食品工业科技(2014年21期)2014-03-11
