
医疗器械洁净环境控制中尘埃数测试的调研分析
2022-05-18 02:03李婧张佳伟李军李臣
医疗装备 2022年7期
李婧,张佳伟,李军,李臣
1 北检润和(北京)技术服务有限公司 (北京 101111);2 北京弘海微创科技有限公司(北京 102308)
洁净室(区)是对尘埃和微生物污染规定需进行环境控制的房间或区域,是无菌、植入医疗器械生产的必备条件[1]。针对不同的生产工艺要求,需要配备相应级别的洁净室(区),这是保证无菌、植入医疗器械质量的重要前提。
为了深入了解医疗器械生产企业对洁净室(区)质量控制的水平,本研究对27家医疗器械生产企业洁净室(区)的日常检测记录进行了调研,重点对生产企业尘埃数日常检测记录中的问题进行了分析总结,并给出了尘埃数规范测试的方法。
1 洁净室(区)洁净度级别的划分
1.1 空气洁净度的发展
世界上最早关于空气洁净度的标准是1963年美国发布的FS209[2],此标准是第1次按照空气中悬浮粒子量的不同对空气级别进行了划分,但标准中只涉及浮游粒子的污染量控制,不涉及微生物污染量的控制。1967年,美国国家航空航天局(National Aeronautics and Space Administration,NASA)标准中关于空气级别出现了100、10 000、100 000级的概念,此概念至今仍是国际生物洁净室级别划分的基准。
在美国的影响下,世界各国相继颁布了本国的空气等级划分标准,各国的级别名称和控制限度虽有所不同,但划分的依据均是基于空气中的尘埃数。
1.2 我国空气洁净度的相关标准
我国最早的关于洁净技术的标准是1979年3月由国家建委科教局批准实施的《空气洁净技术措施》,此标准按每升环境空气中所含尘埃数不同将洁净室分为5个级别,分别为3、30、300、3 000、30 000级。
我国医药领域最早关于洁净技术的标准为《药品质量管理规范》。现行有效的《药品生产质量管理规范》是根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》制定的,于2010年10月19日由原卫生部部务会议审议通过,以2011年1月17日原卫生部令第79号发布,自2011年3月1日起施行[3]。为加强对医疗器械的监管,提升医疗器械生产企业的质量管理水平,保障医疗器械的安全、有效,国家药品监督管理局于2000年8月18日发布了YY/T 0033-2000《无菌医疗器具生产管理规范》[1],并于2000年9月15日正式实施。
2 尘埃数的要求和测试方法
2.1 尘埃数的要求
YY/T 0033-2000是医疗器械生产企业洁净室(区)必须执行的标准,标准中对不同级别洁净室(区)粒子的要求见表1。
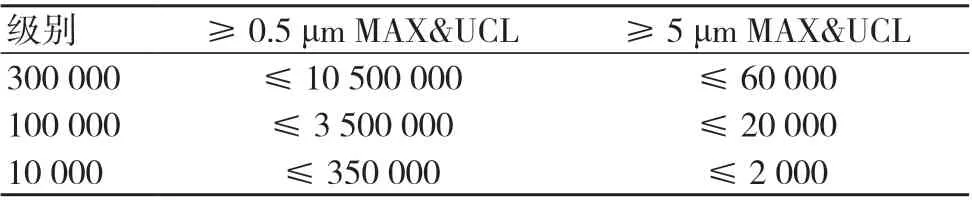
表1 标准YY/T 0033-2000中对洁净室(区)尘埃数的要求(粒/m3)
2.2 尘埃数的判定要求
YY/T 0033-2000中引用的方法标准为GB/T 16292-2010《医药工业洁净室(区)悬浮粒子的测试方法》[4],该标准中的条款7结果判定中明确规定,判断悬浮粒子的洁净度级别应同时满足以下两个条件:a)每个采样点的平均悬浮粒子浓度必须不大于规定的级别界限,即Ai≤级别界限;b)全部采样点的悬浮粒子浓度平均值均值的95%置信上限必须不大于规定的级别界限,即UCL≤级别界限。
3 医疗器械生产企业尘埃数自检记录中存在的问题
本研究对27家医疗器械生产企业洁净室(区)的日常检测记录进行了调研,在调研中发现,仅有4家生产企业尘埃数的测试记录是正确的(正确率不足15%),其他23家生产企业的尘埃数自检记录均存在不同的问题,主要有以下几种。
3.1 判定条件不完整
前述2.2中已经列出标准GB/T 16292-2010中判断悬浮粒子(尘埃数)的洁净度级别应同时满足两个条件,但有21 家生产企业的尘埃数自检记录中均未考虑标准中的条件a),仅对95% 置信上限进行了判定,此判定条件不完整,可能会出现误判。
3.2 采样时间不满足标准要求
很多生产企业使用的粒子计数器的采样流量为2.83 L/min,试验规定的采样时间为1 min,按照此种采样方式,每个采样点的采样量为2.83 L,不满足标准GB/T 16292-2010要求(最小采样量为8.5 L);而若缺少足够的采样量,则不能正确地反映被测房间的粒子浓度。
3.3 未对数据进行统计
在调研的27家生产企业中,有2家生产企业未对粒子的采样数据进行数学统计,而仅罗列、判定了采样的结果。悬浮粒子的测试是基于数学统计的结果进行判定的,我们测试时的采样点存在随机性,此随机性符合正态分布,为了更客观和全面地评估被测房间的悬浮粒子情况,将抽样得到的数据按照95%置信区间进行计算,用得到的数据估计上限表达[3],此结果可更准确地反映被测试房间的悬浮粒子情况,而直接用采样结果进行判定是错误的。
4 尘埃数正确测试举例
4.1 确定采样点数量和位置
图1为检测区域平面示意图,由图可知,微生物室和无菌室的洁净级别为万级,房间面积分别为8 m2和2.7 m2,均小于10 m2,根据GB/T 16292-2010中的最小采样点数目表(表2)可知,本检测房间的采样点数量均为2个;根据标准中给出的布点参考图,对房间中对称的两个点进行取样,采样点位置见图2。
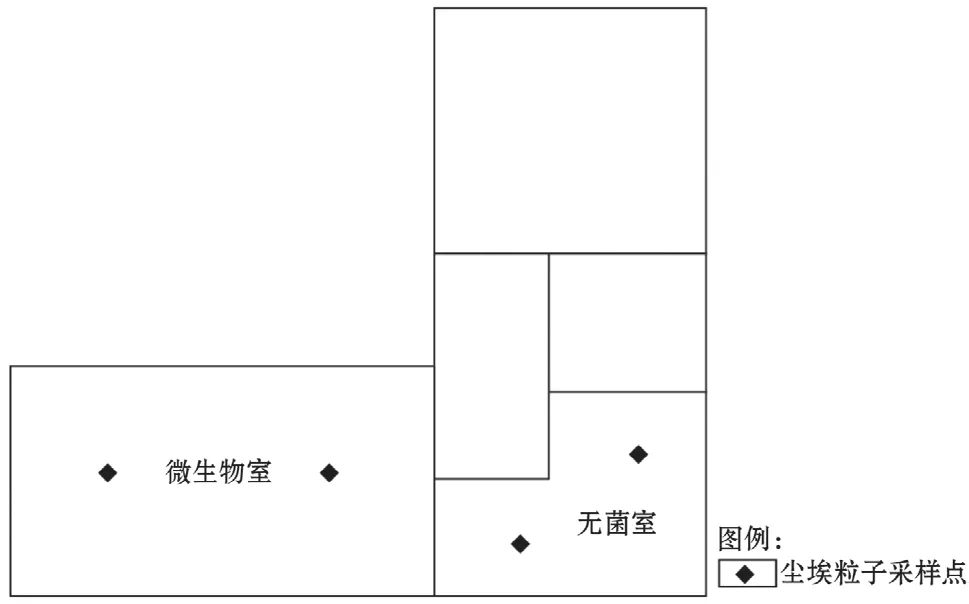
图2 尘埃数采样点布点示意图

表2 尘埃数最小采样点数目(个)
4.2 测试并进行数据分析
根据GB/T 16292-2010中采样次数的要求,使用尘埃粒子计数器对选定的两个采样点进行测试(市场上常见的尘埃粒子计数器的采样量分别为2.83 L/min 和28.3 L/min)。
4.2.1 采样要求
标准GB/T 16292-2010中对采样次数的要求包含两个要点:(1)对于任何被测试的净化区域,采样点数目最少为2个;(2)总采样次数最少为5次,即可在选定的两个采样点上的其中一个采样点上测试2次,另一个采样点上测试3次,或者可选择5个采样点,在每个采样点上测试1次,或者可在一个采样点上测试3次,另一个采样点上也测试3次,总之,只要符合总采样次数不少于5次即可。
4.2.2 确定采样量
标准GB/T 16292-2010中对万级洁净区域最小采样量的要求包含两个要点:(1)粒径≥0.5 μm 的粒子,最小采样量为2.83 L/次;(2)粒径≥5 μm 的粒子,最小采样量为8.5 L/次。为了测试中便于操作,将两个粒径的采样量均定为8.5 L/次,因为标准中的要求为最小采样量,对于粒径≥0.5 μm 的粒子,8.5 L/次的采样量大于2.83 L/次的最小采样量,满足标准要求。
4.2.3 确定采样时间
(1)选用流量为2.83 L/min 的粒子计数器:为了满足8.5 L/次的采样量,在对房间进行尘埃数测试时,每次测试的采样时间最少为3.0 min(8.5 L/次÷2.83 L/min=3.0 min/次)。(2)选用流量为28.3 L/min 的粒子计数器:为了满足8.5 L/次的采样量,在对房间进行尘埃数测试时,每次测试的采样时间最少为0.3 min(8.5 L/次÷28.3 L/min=0.3 min/次),即18 s。
4.2.4 数据统计
使用流量为2.83 L/min 的粒子计数器,根据确定好的采样数量、采样次数、采样量和采样时间,对图1中示意的微生物室和无菌室进行采样,得到以下数据,见表3。
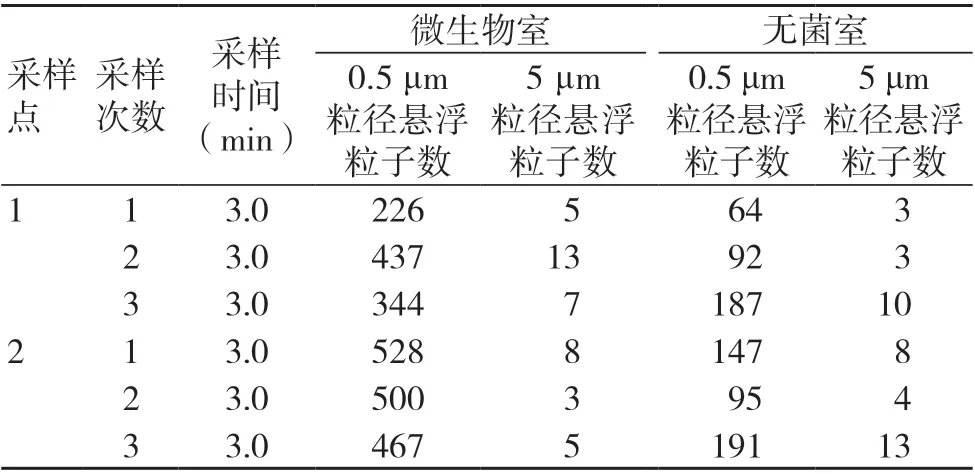
表3 采样数据(粒/8.5 L)
表3中的数据为8.5 L 采样量中含有的粒子数,单位为粒/8.5 L,在对数据进行数学统计前,需要将表3中的数据换算为每立方米中的粒子数,换算步骤如下:(1)1 m3=1 000 L;(2)1 000 L÷8.5 L=118,此为粒/8.5 L 和粒/m3之间的换算系数;(3)1粒/8.5 L×118=1粒/m3;(4)将所有测试得到的数据进行换算,见表4~5。
根据公式(1),计算采样点的平均粒子数,计算结果见表4~5。

式中,M为平均值的均值,即被测区域的平均粒子浓度,单位为粒/m3;Ai为某一采样点的平均粒子数(i=1,2,…,L),单位为粒/m3;L为被测区域内的总采样点,单位为个(本次测试中的L=2)。
根据公式(3),计算标准差,计算结果见表4~5。
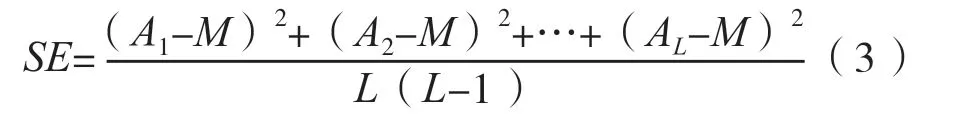
式中,SE为平均值均值的标准误差,单位为粒/m3;M为平均值的均值,即被测区域的平均粒子浓度,单位为粒/m3;Ai为某一采样点的平均粒子数(i=1,2,…,L),单位为粒/m3;L为被测区域内的总采样点,单位为个(本次测试中的L=2)。
根据公式(4),计算95%置信上限,计算结果见表4~5。
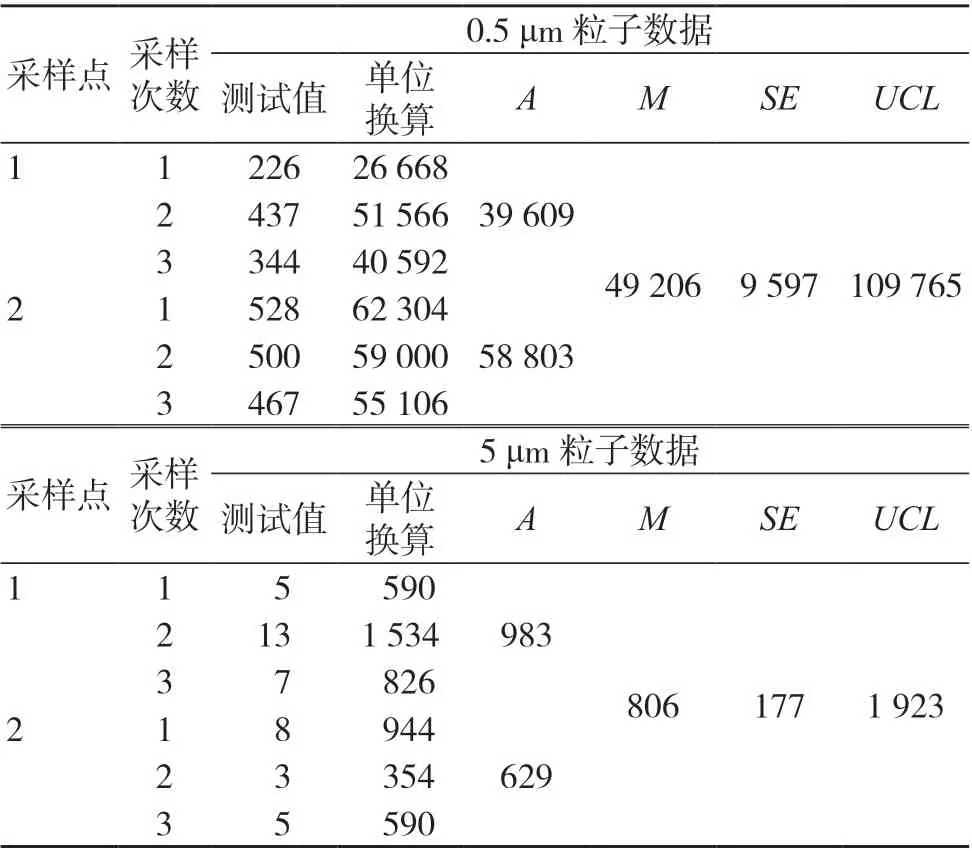
表4 微生物室尘埃数数据统计(粒/m3)

式中,UCL为平均值均值的95%置信上限,单位为粒/m3;M为平均值的均值,即被测区域的平均粒子浓度,单位为粒/m3;SE为平均值均值的标准误差,单位为粒/m3;t为95%置信上限的t分布系数,通过查表6可得,本次测试中的t=6.31。
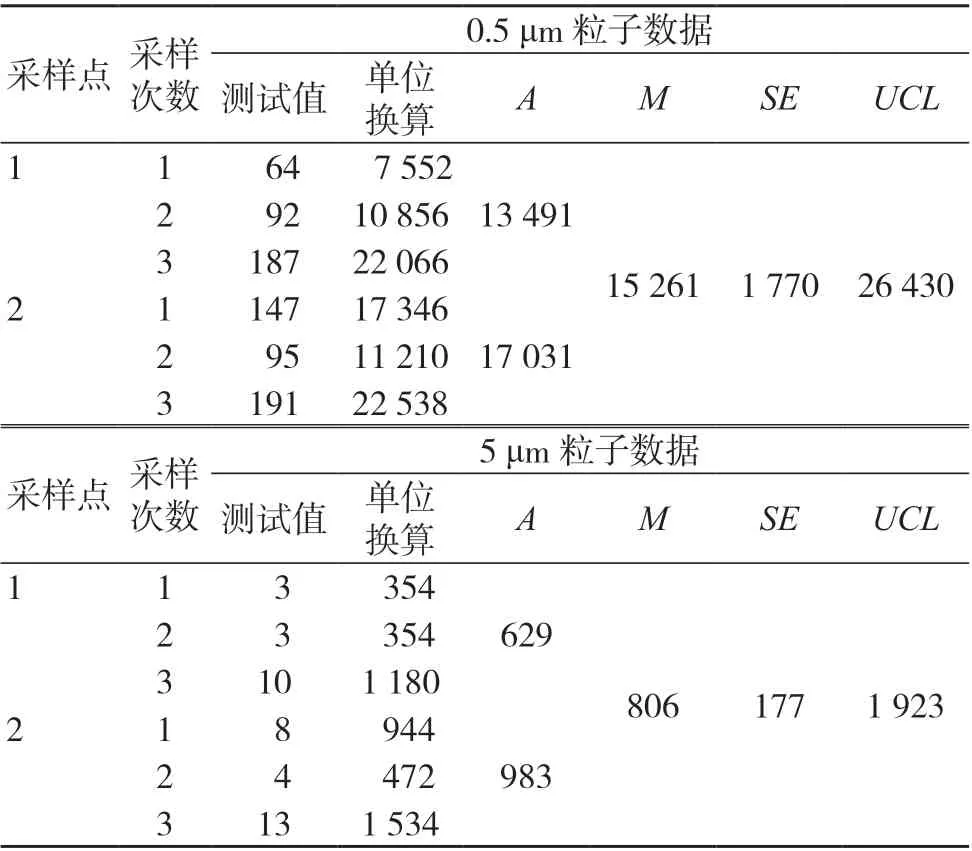
表5 无菌室尘埃数数据统计(粒/m3)
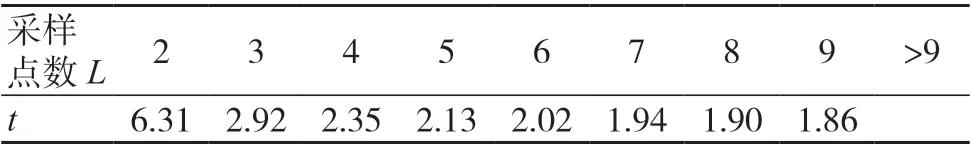
表6 95%置信上限的t 分布系数
4.3 结果评定
根据表4~5中给出的数据,对被测房间微生物室和无菌室的尘埃数进行判定,结果见表7。
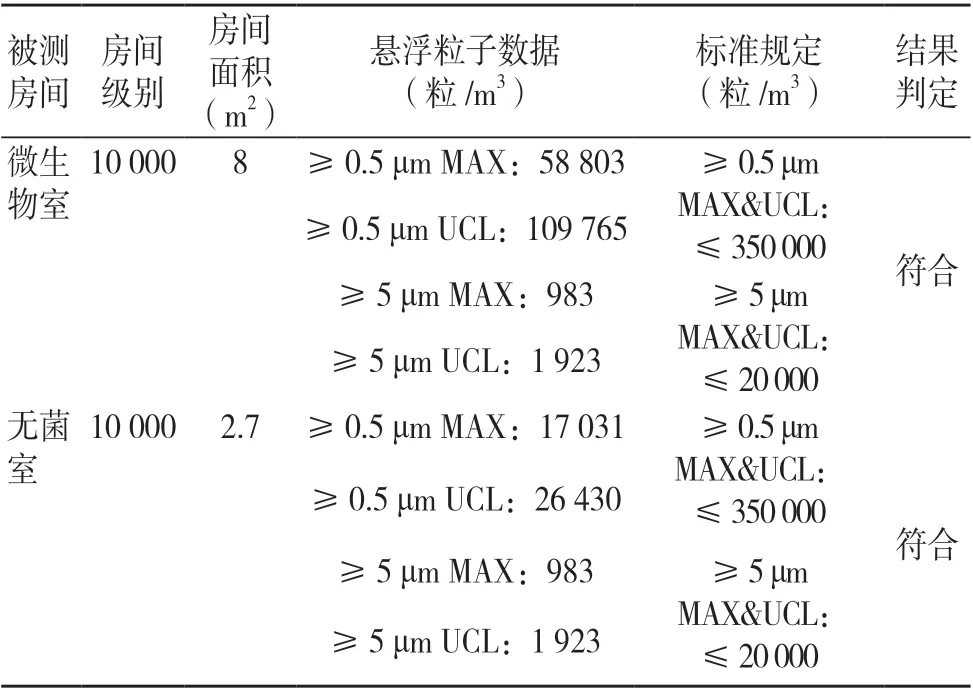
表7 结果判定
5 小结
由表4~5可知,尘埃粒子计数器测量得到的数据与检测项目最终判定时使用的数据相差较大,且两个测试房间不同的测试值经过统计后有可能得到相同的UCL 结果;若不能正确地对测试值进行数学统计,则无法正确判定洁净室(区)的洁净度等级是否满足标准要求。ISO 14644-1:2015《按粒子浓度划分的空气洁净度等级》是世界各国划分空气洁净度等级的依据,我国目前现行有效的洁净室(区)相关受控环境的各项国家标准或行业标准也是直接或间接以此标准为依据进行划分的[5]。由此可见,粒子浓度是划分空气洁净度等级的关键指标,正确对粒子进行检测和报告是医疗器械生产企业对受控环境进行有效控制的重要前提。本研究为生产企业对于尘埃数质量控制提供参考,帮助相关人员正确操作检测设备并进行计算,保证检测结果的准确性,切实保障质量控制工作的可行性、延续性、有效性。
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2022年1期)2022-04-19
西江月(2021年2期)2021-11-24
现代仪器与医疗(2021年2期)2021-07-21
汉字汉语研究(2021年4期)2021-03-09
———占旭刚4
北广人物(2020年48期)2020-12-22
学苑创造·A版(2020年4期)2020-04-24
经济数学(2020年4期)2020-01-15
晚晴(2018年3期)2018-12-06
