
滇黄精叶绿体全基因组序列及其密码子使用偏性分析
2022-06-10 10:30石乃星谢平选李立文国松
热带亚热带植物学报 2022年3期
石乃星, 谢平选, 李立, 文国松*
(1. 云南农业大学农学与生物技术学院,昆明 650201;2. 广东药科大学中药学院,广州 510006;3. 西北大学生命科学学院,西安 710069)
Polygonatum kingianum, also known as jiejiegao and xianren rice in China, belongs to the perennial herb ofPolygonatumMill in Asparagaceae. Its wild resources are widely distributed in southwest China.In addition, it is engaged in relevant artificial cultivation and planting industries in Yunnan, Guizhou,Sichuan[1-3].Polygonatum kingianumis one of the source plants of Rhizoma Polygonati which has high medicinal and edible value[4]. As a medicinal plant,P.kingianumis recorded in various national medical books. Chinese ancient medical book (Ming Yi Bie Lu,Han Dynasty, 220—450 AD) listed Rhizoma Polygonati as the top grade. Modern pharmacological studies have found their main chemical components have antiaging, anti-tumor, immune enhancement, sterilization and anti-inflammatory effects[5].
As an essential subcellular organelle of plants and algae, previous study has found that chloroplast is not only the main place for photosynthesis, but also participates energy transformation[6]. In addition, chloroplasts also have relatively independent genome. In most angiosperms, the chloroplast genome belongs to the maternal inheritance, which has the characteristics of stable structure, conserved coding region sequence,rich information[7-8]. The complete chloroplast genome has been widely used in plant system evolution[9-11],related species identification and genetic diversity analysis[12], chloroplast genetic engineering, etc[13].
Codon, also known as triplet code, as a bridge connecting nucleic acid and protein[14], is the basic unit of biological genetic information transmission. In the case of mutation pressure, natural selection and genetic drift, prokaryotic and eukaryotic organisms generally tend to use one or more specific synonymous codons called synonymous codon usage bias(CUB)[15]. Through the analysis of species codon usage bias, the optimal codon can be determined, which can improve the efficiency and accuracy of related gene expression products, infer the function and expression mode of unknown genes, and provide scientific basis for exploring species relationship and genetic evolution[16-17].
Until now, the codon usage bias of some species has been analyzed[18-20], but the research on the codon usage preference ofP.kingianumhas not been reported yet. In the present study, Illumina Hiseq was used technologies for complete chloroplast genome sequence ofP. kingianum. Based on this, we analyzed the sequence characteristics and codon usage bias of chloroplast genome. It was of great significance to provide a scientific reference for the application and investigation of the chloroplast genome inP. kingianum.
1 Materials and methods
1.1 Material collection and sequencing
Polygonatum kingianumwas collected in Tengchong City of China. The total DNA was extracted from 100 mg of fresh and healthy leaves using the modified CTAB method. Then the complete cp genome was sequenced by using Illumina Hiseq 2000 sequencing platform. The reference specimen (Ji et Wang 2) was deposited at the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences.
1.2 Plastome assembly, annotation, and comparison
First, we assembled the complete chloroplast genome with a reference-based assembly strategies with GetOrganelle using the complete chloroplast genome sequence ofP. kingianum(NCBI reference sequence:MN934979) as reference. Then, the assembly was edited and annotated according to the reference in Geneious V 10.2[21]. We generated a physical map of the cp genome using Organellar GenomeDRAW[22]. Finally,the complete chloroplast genome ofP. kingianumwas submitted to the NCBI (Accession: MW788495).
1.3 Microsatellite analysis
Perl scripts from MISA were used to perform SSR identification with the default parameters (http://pgrc.ipk-gatersleben.de/misa/). The identification criteria were as follows: mono-nucleotide repeat motifs with at least 10 repeats, di-nucleotide repeat motifs with 5 repeats, trinucleotide repeat motifs with four repeats,tetra-, penta- and hexa-nucleotide repeat motifs with three repeats. Compound SSRs were defined as those with a<100-nt interval between two repeat motifs[23-24].
1.4 Phylogenetic analysis
In order to explore the evolutionary relationships ofP. kingianum, the whole chloroplast genome sequences of two genera ofHeteropolygonatumandPolygonatumfrom NCBI along with the obtained chloroplast genome sequence in the present study,were analyzed for phylogenetic analysis. All of the chloroplast genome sequences were aligned with MAFFT[25]implemented in Geneious (10.0.5), and a maximum-likelihood phylogenetic analysis was performed in RAxML[26]under the GTR-GAMMA model with 1 000 bootstrap replicates.
1.5 Codon composition and optimal codon analysis
Based on the consideration of reducing sample error and accurately counting the number of effective codons, we eliminated the sequences with length less than 300 bp, duplicate genes, and coding sequences(CDS) containing stop codon. Fifty-three CDS sequences (start codon: ATG; stop codons: TAA, TAA,TGA and TAG) of the chloroplast genome ofP.kingianumwere used as research samples to analyze the CUB ultimately[27]. In order to analyze the rule of gene base composition, Codon W 1.4.2 software was used for the analysis relative synonymous codon usage (RSCU). We also used the CUSP and CHIPS models in the online software EMBOSS to analyze the GC content of the first base of codon (GC1), the second base of codon (GC2), the third base of codon (GC3), total GC content (GC), and effective number of codons (ENC).The Pearson correlation analysis of the above parameters was carried out using SPSS 24.0 software.
The RSCU value of codon more than 1 are determined to be high frequency codons[28]. Then taking an ENC as the preferred standard, five genes with the highest ENC values and the lowest ENC values in 51 chloroplast genes were regarded as the high and low expression groups. The RSCU values of 2 datasets were calculated and compared byΔRSCU (RSCU in high and low expression groups). The codons satisfying bothΔRSCU>0.08 defined as high expression codons.Finally, by combining high frequency and high expression codons, the optimal codons was defined for the chloroplast genome ofP. kingianum[29].
1.6 Neutrality plot analysis
A neutral graph was drew to research the influence of mutation pressure and natural selection on the chloroplast codon usage pattern ofP. kingianum. GC3values were regarded as abscissa; the average values of GC1and GC2of each gene were seen as GC12, which were ordinate. The correlation analysis of GC3and GC12will be helpful to make scientific judgments on the main factors affecting codon preference[30]. If there is a significant correlation between the two data and the regression coefficient is close to 1, it means that the codon preference is mainly affected by mutation pressure.On the contrary, it indicates that base composition preference is mainly affected by selection pressure.
1.7 ENC-GC3s plot analysis
The effective number of codon (ENC) is a measure of the degree of species independent synonymous codon bias in genes. Its value ranges from 20 to 61,which is negatively correlated with the CUB[31].
ENC-plot analysis can intuitively judge gene codon preference factors. With the GC3s values as horizontal ordinate and ENC values as longitudinal coordinate, two-dimensional scatter plot was drawn[31].Standard curve formula: ENCexp=2+GC3s+29/[GC3s2+(1-GC3s)2][32]. This curve shows the functional relationship between ENC and GC3s only under mutation pressure conditions. The ENC ratio distribution can quantify the results obtained by ENC-plot and clarify how far away or close each gene point is from the curve. The expression∶ENC ratio=(ENC expected value-ENC actual value)/ENC expected value[33].
1.8 Analysis of PR2-bias plot
Based on the analysis of the composition of the four bases (A, T, C, G) at the third position of the chloroplast genome of theP. kingianum, we used G3/(G3+C3) and A3/(A3+T3) as the horizontal and vertical coordinates for analysis. The PR2-bias plot, which analyzes the nucleotide compositions at the third position of codons, are usually used to estimate the effects of mutation pressure and natural selection by analysing the AT bias and GC bias[34]. Through the vector emitted by the center of the plane, we can judge the degree and direction of the 4 kinds of bias[35].
2 Results
2.1 Characteristics of complete chloroplast genome sequence
The length of the complete chloroplast genome sequence ofP. kingianumwas 155 852 bp, with GC content of 37.7%. It contained a pair of inverted repeats (IRs, 26 347 bp each), a small single copy(SSC, 18 525 bp) and large single copy region (LSC,84 633bp). The average GC content of IR, LSC and SSC regions were 43%, 31.6% and 35.7%, respectively.Annotation results showed that there were 132 genes in the chloroplast genome inP. kingianum, including 85 protein-coding genes, 38 tRNA genes and 8 ribosomal rRNA genes. The GC contents of these three types of genes were 38.1%, 53.2% and 55.3%,respectively. The coding region of the gene was 90 607 bp, accounting for 40.3% of the entire chloroplast genome (Fig. 1). The coding genes families ofP.kingiantumchloroplast were involved in four aspects:photosynthesis, self-replication, biosynthesis and unknown function. Table 1 shows the gene functions and groups in the cp genome. Compared with the chloroplast genomes of other species inPolygonatumgenus, such asP. zanlanscianense(155 609 bp)[36]andP. humile(156 082 bp)[37], all cp genomes shared the same gene order and structure, which displayed a high degree of similarity.
2.2 SSR analysis of chloroplast genome
With MISA analysis, a total of 69 SSRs were identified in the chloroplast genome ofP. kingianum.Examination of all SSR loci in the genome showed that the majority of the SSR loci were located in the LSC region, with a number of 50 (72.46%). There were 11 (15.94%) located within the SSC region and the least number of SSRs located in the IR region,with only 8 (11.59%) (Fig. 2). The types of SSRs differed greatly in the number of repeats. The number of mononucleotide repeats was the largest, with 43,and all repeating units were A/T. There were 15, 5,10, and 2, dinucleotide, trinucleotide, tetranucleotide, and pentanucleotide repeats, respectively; while hexanucleotide repeats were not observed. In terms of SSR repeat unit types, tetranucleotide repeat units were the most common, followed by the dinucleotide, trinucleotide repeat units and, finally, the mononucleotide, pentanucleotide repeat units (Table 2).

Table 1 List of identified genes in cp genomes of the Polygonatum kingianum

Table 2 Repeat type, number and frequency of SSRs in complete chloroplast genome of Polygonatum kingianum
2.3 Phylogenetic analysis
The phylogenetic graph revealed that most of species ofPolygonatumwere clustered into a monophyletic clade with a high bootstrap value (Fig. 3). It indicated that the resulting phylogenetic tree we constructed was relatively robust. The sequence ofP.kingianumin this study andP. huanum(P. huanumis a synonym ofP. kingianum) from GenBank clustered together with a support rate of 100%. It showed that the conclusion of morphological identification was supported by molecular evidence. These samples were clearly divided into three branches. Branch I included 8 species (P. urceolatum,P. punctatum,P. stewardtianum,P. oppositifolium,P. tessellatum,P. huanum,P.kingianum). Branch II composed 5 species (P. yunnanense,P. arisanense,P. humile,P. biflorum,P. cyrtonema). The other two species (Heteropolygonatum altelobatumandH. ginfushanicum) were classified into branche III. The verticillate leaf type ofPolygonatumare clustered in a clade, and alternate phyllotaxis species are clustered in anther clade.

Fig. 1 Gene map of the chloroplast genome of Polygonatum kingianum

Fig. 2 Distribution of various types of SSRs in LSC, SSC and IR regions Polygonatum are clustered in a clade, and alternate phyllotaxis species are clustered in anther clade.

Fig. 3 Phylogenetic tree constructed using the maximum likelihood method based on chloroplast genome sequence
2.4 Codon usage bias of chloroplast genome
2.4.1 Codon composition and optimal codon
From the chloroplast genome ofP. kingianum, 51 CDSs suitable for analysis of CUB were selected (Table 3). The average GC content (GCall) of these 51 CDs sequences was 38.28% (30.77%-45.08%). The GC contents at different positions of the codons varied, with the average GC contents at the first position (GC1) being 46.84% (31.79%-58.54%), the second (GC2) being 39.38% (26.61%-55.40%), and the third (GC3) being 28.61% (20.90%-37.23%), indicating that the codons in the chloroplast genome ofP. kingianumpreferred to end with A or U. The statistics of the ENC values of 51 CDs sequences (Table 2) showed that the average value of the ENC of all genes was 48.21 (range from 39.83 to 60.11),suggesting a weak codon preference.

Table 3 ENC value and GC content in different positions of codons in chloroplast 51 CDS of Polygonatum kingianum
Pearson correlation analysis showed that the average GC content (GCall) at all codon locations was significantly correlated with GC1, GC2and GC3. GC1was significantly correlated with GC2, but neither of them reached a significant level with GC3, showing that the composition of the first and second base of the codon was similar, and there was a great difference from the third base. The correlation of ENC with GC1and GC2did not reach significance, but it was highly significant with GC3, indicating that the third base composition can significantly influence codon usage bias (Table 4). The correlation coefficient between ENC and codon counts(CC) was 0.079, which did not reach a significant level,indicating that the CC has a very weak influence on ENC.That is, in this study, the effect of gene length on codon bias analysis was not significant.
There were 30 codons (RSCU>1) in the chloroplast genomic protein coding sequence ofP. kingianum,of which only one codon ended with G or C, and the remaining 29 codons ended with A and U, indicating that the chloroplast genome ofP. kingianumprefers to use Codons ending with A or U; there were 31 codons (RSCU<1), of which 28 ended with G and C, and 3 ended with A and U (Table 5), manifesting that the frequency of codons ending in C and G was relatively low in the chloroplast genome ofP. kingianum.According toΔRSCU values of codes in highand low libraries, 26 highly-expressed superior codon were screened out from the chloroplast genome ofP.kingianum(Table 6). Combining the high expression superior codons with the 30 high-frequency codons described above, nine common codon (UUU, CUU,UCA, CCA, CAU, CAA, AAU, GAU, GGA) were finally identified as the optimal codons for the genome ofP. kingianum, with 4 codons ending with A, and the remaining 5 codons all end with U, whereas the chloroplast protein encoding genome ofP. kingianumpreferred the codons ending with A and U, especially the codons ending with U.

Table 4 Correlation analysis of each gene’s related parameters of Polygonatum kingianum

Table 5 RSCU analysis of CDS in Polygonatum kingianum

Table 6 Optimal codons in chloroplast genome of Polygonatum kingianum
2.4.2 Neutrality plot
To estimate the extent of mutation pressure as well as natural selection contributed to the CUB ofP.kingianum, a neutrality plot was constructed based on the GC12and GC3(Fig. 4). The range was 0.328 1-0.557 6 and 0.209 0-0.372 3 for GC12, GC3, respectively. As shown in Fig. 4, most of the points represented by each gene were distributed above the diagonal of the neutrality plot. Only theycf2 gene was close to the diagonal. The Pearson correlation coefficient between GC12and GC3was 0.142 (P=0.311>0.05),showing that the correlation between them was not significant. The regression coefficient observed was closer to zero (The slope of the regression line was 0.161 1), which inferred that the GC content in the chloroplast genome ofP. kingianumwas highly conserved. Natural selection played a remarkably important role in the CUB ofP. kingianum. And the mutation pressure accounted for a minority of the affecting factors.
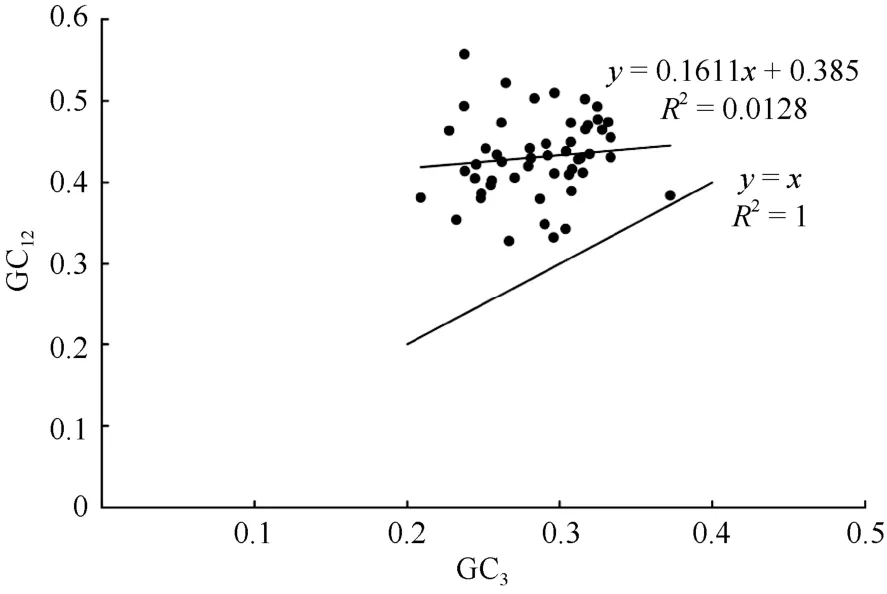
Fig. 4 Neutrality plot analysis
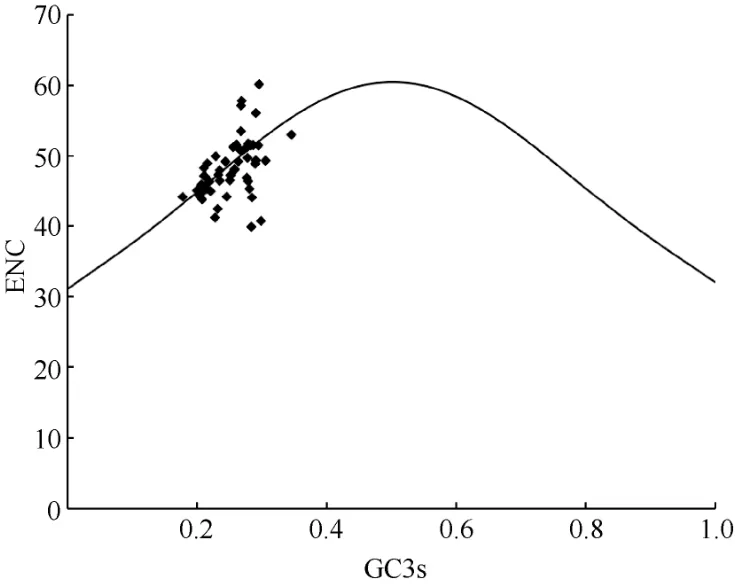
Fig. 5 Analysis of ENC-plot
2.4.3 ENC-GC3s plot
The standard curve in the ENC-plot reflected the relationship between ENC and GC3s only when the influence of selection pressure is excluded. Figure 5 showed that some genes in the chloroplast ofP.kingianumwere located near the standard curve. The actual ENC value of this part of the gene was close to the expected ENC value, indicating that the mutation effect was greater than natural selection; while the position of the other part of the gene farther from the standard curve represented natural selection factors were stronger than mutations. In order to quantify the closeness of genes to the standard curve, the ENC ratio was used to count the frequency of ENC ratios(Table 7). The results showed that there were only 21 genes with ratios in the range from -0.05 to 0.05,accounting for 41.18% of the total number of genes.That means, most genes were far away from the standard curve, and the codon preference was related to the difference of GC3s, indicating that the codon preference of the chloroplast genome ofP. kingianumwas more affected by selection than mutation.
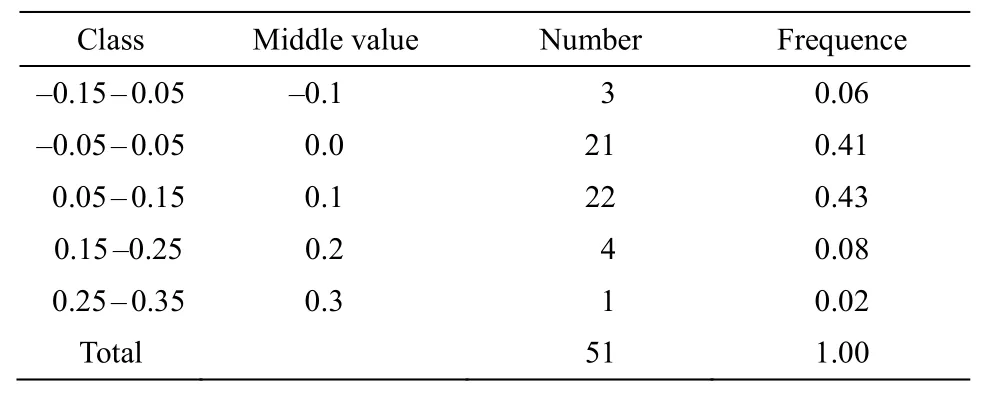
Table 7 Distribution of ENC ratio
2.4.4 PR2-plot
PR2-plot analysis showed that the chloroplast genes ofP. kingianumwere scatteredly distributed in the four regions of the chart, and most of the genes were distributed in the lower left part of the chart(Fig. 6), indicating that the frequency of T base in the third codon was higher than that of A base, and that of C base was higher than that of G base, that is, the frequency of pyrimidine was higher than that of purine.If the codon usage pattern is completely caused by mutation, the usage frequency of the four bases should be equal. The biased usage of four bases indicated that the usage pattern of chloroplast codon inP. kingianumwas not only influenced by mutation, but also by other factors, such as selection pressure.

Fig. 6 Analysis of PR2-plot
3 Conclusion and discussion
Polygonatum kingianumis one of medicine and food homologous plants announced by the National Health Commission, PRC. For thousands of years, its medicinal effect and edible value has been widely recognized by Chinese people. Up to date, it has integrated medicinal, edible, ornamental and health function which has extremely high economic value and social benefits. In summary,P. kingianumhas good development and research prospects[2].
In this study, basic research on the chloroplast ofP. kingianumwas conducted. The assembly annotation results showed that the chloroplast genome ofP.kingianumwas 155 852 bp in length, including one large single-copy region, one small single-copy region and two inverted repeat regions, which is consistent with the typical tetrad structure of the chloroplast genome of most angiosperms[38]. The study found that the chloroplast genome ofP. kingianumwas not much different from the chloroplast genome of other species in the genusPolygonatumin sequence length. Simple sequence repeats (SSRs) are an important part of the plant chloroplast genome which play an indispensable role in gene expression, transcription regulation, chromosome construction, and physiological metabolism[39].After statistical analysis of MISA online software, 69 SSR loci were detected. These SSR loci will be helpful for subsequent research on the population genetics ofP. kingianum. Studies have shown thatP. kingianumis a species with large morphological variation, and it is difficult to identify this species only from morphology. The phylogenetic tree results of this study provide reliable DNA molecular evidence support for the morphological identification ofP. kingienum. Compared with other plants ofPolygonatumgenus, the relationship betweenP. kingianumandP. tessellatumis closer, and the geographic distribution of 2species is basically the same.
Although CUB is affected by many factors,natural selection and mutation are key factors that affect codon usage preference[40]. The results of codon bias in the chloroplast genome ofP.kingianumshowed that the average effective codon ranged from 39.830 to 60.106, which suggested a weak codon preference among these chloroplast genes.
The results of related parameters analysis and neutrality plot showed that the third position of codon had low base composition similarity with the first and second position. The correlation between the first position and the other two positions of the codon was not significant, indicating that the codon bias was subject to a strong degree of selection.
The results of ENC-plot analysis also confirmed the above argument. The ENC-plot graph showed that only a small part of genes distributed near the standard curve, and the actual ENC value of these genes were basically consistent with the theoretical ENC values.It indicated that these codon preferences were greatly affected by mutations. The scattered points of most genes were far from the standard curve. The actual ENC value of this part of the gene was quite different from the theoretical ENC value, indicating that it was more easily affected by the selection.
The analysis result of PR2-plot concluded that at the third codon position of cp genes, pyrimidines (C and T) were used more frequently than purines (A and G), which verified that the codon usage pattern of the chloroplast genes ofP. kingianumwas more affected by selection factors.
Based on the results of the high-frequency and highly expressed codons, nine codons (UUU, CUU,UCA, CCA, CAU, CAA, AAU, GAU, GGA) were obtained as the optimal codons ofP.kingianumchloroplast genes finally. This preference pattern is consistent with the results of codon bias analysis of chloroplast genes inOryza[41],Oncidium gower[42],Panicum miliaceum[43], etc. These indicate thatP.kingiantumprefers to use codons ending in AT like other monocotyledonous plants.
The chloroplast genome sequences are not only most valuable for understanding plant evolution and phylogeny but also have made some achievments in chloroplast transformation technology. However, chloroplast genome has rarely been used in evolution and phylogeny of theP.kingiantum.Only three relevant research have been repored[37,44-45]. These reports mainly discussed the phylogenetic relationship and identification ofPolygonatumgenus. Up to now, chloroplast genetic transformation technology has been applied in a variety of plants. Degray, et al[46]transferred the antimicrobial peptide geneMSI-99into the chloroplast genome of tobacco by chloroplast transformation technology, and the descendants of transgenic plants showed high antibacterial activity.Chakrabarti, et al[47]incorporated a truncatedBacillus thuringiensis cry9Aa2gene in the plastid genome of tobacco to control ofpotato tuber moth (Phthorimaea operculella). But, it has not been found any studies on chloroplast genetic transformation ofPolygonatumspecies by far.
In conclusion, we sequenced and analyzed the complete cp genome ofP. kingiantum, which exhibits conserved structure. The phylogenetic relationship based on the plastid genome data shows that the chloroplast genome as ultra-barcoding has great potential in the identification ofPolygonatumspecies.Comprehensive analysis found that the codon bias of theP. kingiantumchloroplast genome is weak, and the factors that affect the formation of codon bias in the chloroplast protein-coding genes ofP. kingianumdo not depend on a single factor, but are the result of mutations, selection and many other factors. According to the codon usage characteristics of its chloroplast genome, we screened out 9 optimal codons for the chloroplast genome ofP. kingiantum. The current study provides a scientific reference for the identification of germplasm resources, genetic breeding as well as for prediction of unknown functional genes,discovery of new genes, improvement of foreign gene expression inP. kingianum.
猜你喜欢
新农业(2022年16期)2022-11-07
声屏世界(2022年13期)2022-10-08
小小说月刊(2022年14期)2022-07-18
中老年保健(2021年11期)2021-08-22
福建农业学报(2021年6期)2021-08-18
婚育与健康(2020年12期)2020-02-26
校园英语·下旬(2019年11期)2019-11-28
发明与创新·中学生(2019年6期)2019-06-26
生物学教学(2018年2期)2018-08-07
安徽农业科学(2018年1期)2018-05-14
