
尼拉帕尼关键中间体的合成工艺改进
2022-07-06 14:27于立国
上海医药 2022年11期
摘 要 本研究报道了一条尼拉帕尼关键中间体(S)-叔丁基3-(4-氨基苯基)哌啶-1-羧酸(1)的新合成路线。以3-溴吡啶作为原料,经过熊田偶联、催化氢化、拆分、硝化、还原、Boc保护制得1。采用镍催化的熊田偶联替代原来钯催化的Suzuki偶联,不仅大幅降低了成本,同时还提高了收率;用Pd/C催化氢化替代PtO2催化,Pd/C催化剂可实现多次套用活性不下降,显著降低了成本;将氨基放在拆分之后引入,可以将拆分收率提升一倍,同时提高了产品的ee值。总收率17.5%,产品纯度98.95%,手性纯度99.92%。
关键词 尼拉帕尼 产业化 合成
中图分类号:O626.322 文献标志码:A 文章编号:1006-1533(2022)11-0064-04
引用本文 于立国. 尼拉帕尼关键中间体的合成工艺改进[J]. 上海医药, 2022, 43(11): 64-67.
Improvement of synthetic process for key niraparib intermediate
YU Liguo
(Changzhou Pharmaceutical Factory, Shanghai Pharmaceutical Group, Changzhou 213018, China)
ABSTRACT A novel route for the synthesis of key intermediate (S)-tert-butyl 3-(4-aminophenyl)piperidine-1-carboxylate(1) was established. Using 3-bromopyridine as raw material, 1 was prepared by Kumada coupling, catalytic hydrogenation, separation, nitrification, reduction and Boc protection. The Kumada coupling catalyzed by nickel was used instead of palladiumcatalyzed Suzuki coupling, which can not only greatly reduce the cost, but also improve the yield. Using Pd/C catalytic hydrogenation instead of PtO2 catalyst, the Pd/C catalyst can achieve repeated application without activity decrease, and the cost can be significantly reduced. Adding the amino group after the resolution can double the yield and increase the ee value of the product with total yield 17.5%, product purity 98.95% and chiral purity 99.92%.
KEY WORDS niraparib; industrialization; synthesis
尼拉帕尼(niraparib,商品名Zejula)是一种口服小分子PARP抑制剂[1],用于治疗卵巢癌,由Tesaro医药公司开发,先后于2017年3月27日和2017年11月16日在美国和欧洲获批上市。卵巢癌是婦女中一种常见的癌症[2-3],也是女性因癌症死亡的第五大常见原因之一,尼拉帕尼是首款无需检查brca突变或其他生物标志物的状况,就能在临床上显著改善复发性卵巢癌患者的无进展生存期的聚腺甘酸二磷酸核糖基聚合酶(poly ADPribose polymerase,PARP)抑制剂[4-5]。此外,它仅需每日口服一次,就能起到控制病情的效果,是一种非常易用的维持疗法。
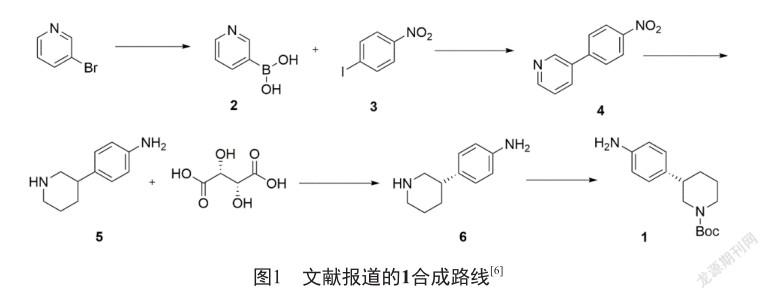
文献报道的(S)-叔丁基3-(4-氨基苯基)哌啶-1-羧酸(1)的主要合成路线如图1所示[6],以3-溴吡啶作为起始原料,经过硼酸化制得3-硼酸吡啶(2)[7],2与4-硝基碘苯(3)经过Suzuki偶联[8]反应制得3-(4-硝基苯基)-吡啶(4),偶联用到昂贵的四三苯基膦钯催化剂以及3,同时反应收率仅有60%,成本很高。4经过催化氢化制得4-(哌啶-3-基)苯胺(5)[9],PtO2非常昂贵,且较难高效回收。5经过L-酒石酸拆分制得(S)-4-(哌啶-3-基)苯胺(6),此步拆分收率仅20%,并且ee值仅能达到80%,不仅物料损失严重,还给后续纯化带来很大压力。最后,经过Boc保护制得1 [9]。
1是合成尼拉帕尼的关键中间体,鉴于尼拉帕尼巨大的药用前景,开发一条环境友好、成本低、适合产业化的合成路线变得尤为重要。
1 材料与方法
1.1 仪器
LC-20AD高效液相色谱仪(日本岛津公司);ZF-8型暗箱式四用紫外线分析仪(上海嘉鹏科技有限公司);GF254薄层层析(TLC)板;P6107-C电子天平(阿拉丁公司)。
1.2 试剂
3-溴吡啶及其他试剂均为市售工业级商品。
1.3 方法
1.3.1 化合物1新合成路线
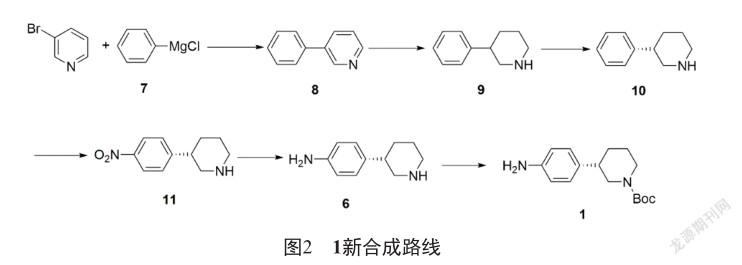
本研究开发了一条环境友好、成本低、适合产业化的新合成路线(图2)。该路线以3-溴吡啶作为起始原料,与苯基氯化镁(7)进行熊田偶联制得3-苯基吡啶(8),再经过催化氢化反应制得3-苯基哌啶(9),9经过L-酒石酸拆分制得(S)-3-苯基哌啶(10),再经过硝化反应制得(S)-3-(4-硝基苯基)哌啶(11),11经过还原反应制得(S)-4-(哌啶-3-基)苯胺(6),最后,经过Boc保护制得1。
1.3.2 HPLC检测
1)有关物质 采用峰面积归一化法:Waters XBridge phenyl柱(4.6 mm×150 mm,3.5 μm);流动相A为0.02 mol/L乙酸铵-乙腈=100∶5,B为乙腈,梯度洗脱(0~40 min:A 95%;40~50 min:A 45%;50~60 min:A 20%;60~70 min:A 95%);柱温30 ℃;流速1.0 mL/min;检测波长235 nm;进样量10 μL;保留时间35.1 min。
2)手性 采用峰面积归一化法:大赛璐OJ-RH柱(4.6 mm×250 mm,5 μm);流动相A为水,B为乙腈,梯度洗脱(0~30 min:A 60%;30~40 min:A 20%;40~50 min:A 60%);柱温35 ℃;流速0.7 mL/min;检测波长235 nm;进样量10 μL;保留时间29.2 min。
2 结果
2.1 化合物8制备
向3 000 mL反应瓶中,投入3-溴吡啶(80 g,506.3 mmol)和四氢呋喃800 mL,通氮保护,冷却反应液至0 ℃;然后,于0~5 ℃下加入1,2-双(二苯基膦)乙烷氯化镍(1.87 g,3.5 mmol),保温搅拌10 min左右;再于0~5 ℃下滴加苯基氯化镁(2 mol)354 mL,滴加时间约2 h,滴加完毕,于0~5 ℃保温反应3~4 h,TLC检测控制至原料3-溴吡啶转化完全;向反应液中慢速加入水800 mL淬灭反应,再加入醋酸异丙酯500 mL,搅拌15 min,静置分层得上层有机相;下层水相用醋酸异丙酯500 mL再萃取一次,合并有机相,水相经TLC确认没有产品后,弃去;向合并的有机相中加入3 mol/L盐酸500 mL,搅拌15 min,静置分层得下层水相(产品层),上层有机相再加入3 mol/L盐酸200 mL,搅拌15 min,静置分层得下层水相,合并水相;用40%氢氧化钠水溶液调节合并水相的pH至8~10左右;用甲基叔丁基醚500 mL×2萃取水相,合并有机相,水相复测pH应为8~10左右;用适量无水硫酸钠干燥合并的有机相,同时加入活性炭4 g,搅拌脱色,过滤,用适量甲基叔丁基醚漂洗滤饼,收集滤液;于40~50 ℃水浴减压浓缩滤液至干,得到油状物8(74 g,476.8 mmol),收率94.2%。1H- NMR(400 MHz,CDCl3):δ 8.67(1H,d,J=4.8 Hz),8.01~7.98(2H,m),7.75~7.72(2H,m),7.51~7.46(2H,m),7.43~7.40(1H,m),7.25~7.20(1H,m)。
2.2 化合物9制备
向高压釜中投入8(60 g,386.6 mmol)、乙酸60 mL、甲醇240 mL和Pd/C(5%)12 g,氮气置换三次,氢气置换三次,通氢加压至2 Mpa;加热升温至70 ℃,氢化约24 h,TLC检测,原料转化完全。冷却反应液至10~20 ℃,抽滤,用甲醇60 mL漂洗,收集滤液,回收钯碳;于40~50 ℃水浴减压浓缩滤液至干,冷却至10~20 ℃,加入水300 mL,搅拌溶解,冷却至10 ℃,用适量30%氢氧化钠水溶液调pH至10,再继续搅拌,复测pH至10;加入甲基叔丁基醚300 mL,萃取,分层,水相再用甲基叔丁基醚300 mL萃取一次,分层,合并有机相,水相TLC检测后确保没有产物;向合并的有机相中加入饱和氯化钠300 mL洗涤,分层得上层有机相,用适量无水硫酸钠干燥,过滤,适量甲基叔丁基醚漂洗,收集滤液;于40~50 ℃水浴减压浓缩滤液至干,得油状物9(56 g,347.3 mmol),收率89.8%。1H-NMR(400 MHz,CDCl3):δ 7.37~7.25(m,2H),7.25~7.16(m,3H),3.25~3.01(m,2H),2.77~2.47(m,3H),2.09~1.88(m,2H),1.77(ddd,J=8.6,3.7,2.1 Hz,1H),1.63(dtdd,J=11.5,6.1,4.1,3.0 Hz,2H)。
2.3 化合物10制備
向反应瓶中投入9(517.7 g,3.21 mol)和甲醇3 880 mL,搅拌使溶液澄清,然后缓慢加入L-酒石酸(482.6 g,3.22 mol)和甲醇3 880 mL配成的溶液,过程中可能会有固体析出,加毕,于30~35 ℃搅拌4 h;过滤,收集固体;将湿固体投入反应瓶中,加入甲醇7 L,回流打浆1 h,然后冷却至30~35 ℃,继续搅拌2 h,过程控制温度30~35 ℃;过滤,得湿品,于50 ℃鼓风干燥至恒重,向反应瓶中投入水2 L,搅拌分散使溶液不完全澄清,加入4 mol/L氢氧化钠水溶液661 mL,过程温度控制在20~30 ℃,搅拌15 min,测控pH至12~13。加入醋酸异丙酯1.2 L,搅拌萃取,水相再用醋酸异丙酯600 mL×2萃取,合并有机相,适量无水硫酸钠干燥,过滤,于50 ℃水浴减压浓缩至干,得浅黄色油状物10(199 g,1.23 mol),收率38.4%。1H-NMR(400 MHz,CDCl3):δ 7.37~7.25(m,2H),7.25~7.14(m,3H),3.25~3.01(m,2H),2.76~2.46(m,3H),2.09~1.88(m,2H),1.79(ddd,J=8.6,3.7,2.1 Hz,1H),1.63(dtdd,J=11.5,6.1,4.1,3.0 Hz,2H)。
2.4 化合物11制备
向反应瓶中投入10(300 g,1.86 mol)、冰醋酸750 mL,搅拌使溶液清澈,放热(过程温度可升至50 ℃);冷却反应液至-10 ℃,然后滴加98%浓硫酸186 g和冰醋酸300 mL配成的溶液,过程放热,控制温度≤5 ℃;继续滴加68%浓硝酸211.5 g和冰醋酸300 mL配成的溶液,滴加时间约30 min,过程控制温度≤5 ℃,滴加完毕,继续滴加98%浓硫酸750 mL,过程控制温度≤25 ℃;加热升温至60~65 ℃,保温反应约3 h,TLC检测确认原料转化完全;冷却反应液至20~30 ℃,然后将反应液加入到2 L冰水中,过程控制温度≤5 ℃,然后缓慢加入氢氧化钠2 438 g和水7.6 L配成的氢氧化钠水溶液,过程温度不超过40 ℃;然后加入醋酸异丙酯2 L,萃取,水相再用醋酸异丙酯2 L×2萃取二次,TLC确认水相中无产品,合并有机相,加适量无水硫酸钠干燥,过滤;50 ℃水浴减压浓缩至干得酒红色固体11(308 g,1.49 mol),收率80.3%。
2.5 化合物6制备
向压力釜中投入11(20 g,97.0 mmol)、甲醇100 mL和水100 mL,搅拌至溶液澄清,再投入Raney Ni 4 g,氮气置换3次,氢气置换3次;升温至70 ℃,加压至1.5 Mpa,保温反应4 h,消耗氢气至0.75 Mpa后压力不再下降,继续加压至1.5 Mpa,并在此压力下保温2 h。经TLC检测确认原料转化完全后,出料,抽滤,用适量甲醇漂洗,回收Raney Ni;滤液45 ℃水浴减压浓缩除去甲醇,水相pH用氢氧化钠调至10~12,再加入二氯甲烷100 mL萃取两次,直至水相没有产品残留;有机相用无水硫酸钠干燥,过滤,滤液于45 ℃水浴减压浓缩至干得到粗品;向反应瓶中投入乙酸乙酯35 mL,加热回流,使溶液不完全澄清;加入正己烷350 mL,继续回流30 min,然后降温至5~10 ℃,析晶4 h,过滤,50 ℃鼓风干燥至恒重,得固体6(13.8 g,78.3 mmol),收率80.7%。1H NMR(400 MHz,CDCl3,300 K) δ 6.96(2H,d,J=8.0 Hz),6.60(2H,d,J=8.0 Hz),3.56(2H,br s),3.13~3.09(2H,m),3.13~3.10(3H,m),1.98~1.92(1H,m),1.79~1.75(1H,m),1.66~1.53(2H,m)。
2.6 化合物1制备
向反应瓶中投入6(10 g,56.7 mmol)、二氯甲烷200 mL和三乙胺10 mL,冷却反应液至0 ℃。温度控制0~5 ℃,然后滴加(Boc)2O(11.8 g,54.1 mmol)和二氯甲烷20 mL配成的溶液,滴加约1 h,然后在0~5 ℃保温搅拌2 h左右使溶液澄清;经TLC检测确认原料转化完全后,向反应液中,加入水200 mL搅拌,分层得下层有机相,用水100 mL×2水洗,分层得的有机相用适量无水硫酸钠干燥,过滤,滤液于40~50 ℃水浴减压浓缩至干,得到粗品;向粗品中加入乙酸乙酯14.3 mL,加热至溶液澄清,再加入正己烷143 mL,加热回流至溶液澄清,降温至20 ℃,搅拌析晶2 h,过滤,滤饼于40~50 ℃鼓风干燥至干,得类白色固体1(13 g,47.0 mmol)收率83.0%。1H NMR(400 MHz,CDCl3,300 K)δ 7.03(2H,d,J=8.0 Hz),6.66(2H,d,J=8.0 Hz),4.23~4.06(2H,m),3.64~3.48(1H,m),2.75~2.50(3H,m),2.01~1.92(1H,m),1.77-1.59(1H,m),1.65-1.52(3H,m),1.47(9H,s)。
3 讨论
本工艺与现有文献方法相比,有如下优点:①用镍催化的熊田偶联替代原来钯催化的Suzuki偶联[10],不仅大幅降低了成本,同时将收率从文献报道的79%提高至94.2%[6]。②用Pd/C催化氢化替代PtO2催化,Pd/C催化剂可实现多次套用活性不下降,顯著降低了成本。③将氨基放在拆分之后引入,可以将拆分收率提升一倍,同时提高了产品的ee值。
改进后的工艺,反应条件温和、操作简便、手性纯度高、成本低廉,总收率17.5%,有关物质纯度98.95%,手性纯度99.92%,适合工业化生产。
致谢:感谢浙江大学为本研究的核磁共振检测提供支持。
参考文献
[1] 陈本川. 治疗卵巢癌新药——尼拉帕尼(niraparib)[J]. 医药导报, 2017, 36(10): 1209-1214.
[2] 刘杰, 颜玮, 徐艳, 等. 2015年中国卵巢癌发病与死亡分析[J]. 中国肿瘤防治杂志, 2021, 28(6): 407-411.
[3] 徐杰茹, 陈磊, 王冕, 等. 1990-2019年中国女性卵巢癌发病趋势分析与预测[J]. 现代预防医学, 2021, 48(19): 3457-3460.
[4] 李安沣, 赵冬旭, 高佳音, 等. PARP抑制剂在卵巢癌维持治疗中的规范化应用[J]. 中国医师杂志, 2021, 23(12): 1918-1920.
[5] 韩宇, 赵临襄. 尼拉帕尼(Niraparib)[J]. 中国药物化学杂志, 2017, 27(5): 421.
[6] Wallace DJ, Baxter CA, Brands KJM, et al. Development of a fit-for-purpose large-scale synthesis of an oral PARP inhibitor[J]. Org Process Res Dev, 2011, 15(4): 831-840.
[7] Denton TT, Zhang X, Cashman JR. 5-substituted, 6-substituted, and unsubstituted 3-heteroaromatic pyridine analogues of nicotine as selective inhibitors of cytochrome P-450 2A6[J]. J. Med. Chem, 2005, 48(1): 224-239.
[8] Fu XL, Wu LL, Fu HY, et al. Efficient diphosphane-based catalyst for the palladium-catalyzed Suzuki cross-coupling reaction of 3-pyridylboronic acids[J]. Eur JOC, 2009(13): 2051-2054.
[9] Jones P, Altamura S, Boueres J, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors[J]. J Med Chem, 2009, 52(22): 7170-7185.
[10] Wu ZH, Si TD, Xu GQ, et al. Ligand-free nickel-catalyzed Kumada couplings of aryl, bromides with tert-butyl Grignard reagents[J]. Chinese Chem Lett, 2019, 30(3): 597-600.
猜你喜欢
纺织科学研究(2021年9期)2021-10-14
今日农业(2019年14期)2019-09-18
宝藏(2018年12期)2019-01-29
中国化肥信息(2019年4期)2019-01-17
知识经济·中国直销(2017年7期)2017-07-24
现代商贸工业(2016年14期)2016-12-27
安徽理工大学学报·自然科学版(2016年4期)2016-12-23
考试周刊(2016年85期)2016-11-11
农业与技术(2016年15期)2016-11-09
科技视界(2016年18期)2016-11-03
