
贵金属Pt促进Co基费托合成催化剂的原子尺度结构分析
2022-08-22 10:58韦春洪王盼盼江成发刘岳峰
高等学校化学学报 2022年8期
韦春洪,蒋 倩,王盼盼,3,江成发,刘岳峰
(1.四川大学化学工程学院,成都 610065;2.中国科学院大连化学物理研究所,大连 116023;3.中国科学院大学,北京 100049)
费托合成反应(FTS)可以将天然气、煤炭、生物质以及工业废气CO2转换而来的合成气,转化为甲醇、高级醇、低碳烯烃和清洁燃料等高附加值产品,受到了研究者的广泛关注[1]. 研究发现[2,3],在费托反应生成长链烃类的过程中,CO和H2在活性位点上首先解离形成CHx中间物种,然后CHx中间物种在催化剂表面偶联,形成长碳链的碳氢化合物,使得费托合成的产物分布遵循Anderson-Schulz-Flory(ASF)分布. 费托合成常用的催化剂主要有铁基、钴基和钌基等,其中钴基催化剂具有稳定性好、重质烃选择性高以及水煤气变换反应活性低等优势,得到了广泛应用,也是研究较多的一类费托合成催化剂[4]. 然而,钴基费托催化剂的活性相对较低,钴的储量较少,同时也因为在电池、合金等领域的大量使用而导致价格偏贵,制约了其工业化应用[4]. 因此,通过对钴基费托合成催化剂的改进,有效地提高钴元素的利用效率和费托合成产物的价值一直是研究的重点. 针对这些问题,研究者进行了大量的研究,已经发现粒径[5]、晶相[6~8]、载体[9,10]、助剂效应[11,12]以及特殊空间结构[13]等均会影响钴基催化剂的性能. 与此同时,随着新表征技术的发展,利用新表征技术以及表征方法对费托合成催化剂结构的深入解析,理解其构效关系,也是未来的一个重要研究方向[14~16].
费托反应涉及H—H键的断裂、CO活化以及C—C偶联等过程,其中H—H键的断裂也是影响反应活性的一个关键步骤[17,18]. 钴基催化剂活化解离H2的能力相对来说较弱,可通过添加Pt,Re,Ru等贵金属来提高其费托反应活性[19,20]. 贵金属Pt由于具有优异的H2解离能力,成为钴基费托合成中一种重要的贵金属助剂[19]. 研究发现,Pt 的添加增加了氧化钴的形核中心,在同样钴含量的情况下,形核中心越多,越有利于纳米颗粒的形成,使金属钴的颗粒变小、分散性提高[19]. 贵金属Pt的添加还有助于Co3O4到CoO以及CoO到Co的还原,并且减少硅酸钴、铝酸钴等难还原物种的形成,提高催化剂中钴的还原度,增加暴露出的活性位点的数量[12,21]. 关于Pt的分布和促进机制,Davis等[21]报道Pt倾向于分布在钴纳米颗粒的边缘,并且不形成Pt—Pt键,并认为还原首先在Pt上发生,解离的H溢流到氧化钴上,将氧化钴还原,形成金属钴的颗粒. 然而,对于贵金属Pt在钴基费托催化剂上的分布状态,直观证据还比较少.
多孔碳化硅(β-SiC)具有高导电性及化学惰性,可以排除金属、助剂与载体间相互作用的影响[22].本文以β-SiC为载体,通过共浸渍法制备了Co-Pt/SiC催化剂;借助球差校正的扫描透射电镜,结合程序升温、化学吸附以及同步辐射X射线吸收等实验,对催化剂中贵金属Pt的分布状态以及作用机制进行解析,以期在原子尺度下揭示工业催化剂中Pt助剂的分布,分析Pt对费托合成反应的促进作用.
1 实验部分
1.1 试 剂
六水合硝酸钴[Co(NO3)2·6H2O,纯度98%~102%]和四氨合硝酸铂[Pt(NH3)4(NO3)2,纯度99.99%]均购自阿法艾莎(中国)化学有限公司.
1.2 催化剂的制备
β-SiC 载体购于西卡特公司(SICAT,https://www. sicatcatalyst. com),经过研磨、筛分成160~400 μm,备用. 随后以Co(NO3)2·6H2O 为前驱体,用等体积浸渍法将10%(质量分数)Co 浸渍在合成的β-SiC上. 浸渍得到的固体在室温下老化4 h后,放入110 ℃鼓风烘箱中干燥8 h,以去除多余的水和乙醇,最后用马弗炉在350 ℃下焙烧2 h,得到Co/SiC催化剂.
Co-Pt/SiC 催化剂通过共浸渍法制备. 具体步骤为:首先将Co(NO3)2·6H2O和Pt(NH3)4(NO3)2溶于一定量的去离子水,然后用等体积浸渍法浸渍在β-SiC载体上,经过和Co/SiC相同的老化、干燥和焙烧处理后,得到Co-Pt/SiC催化剂.
1.3 催化剂的表征
X 射线衍射分析(XRD)在荷兰PAN analytica 公司的X'pert-Pro 衍射仪上进行,以CuKα为射线源,扫描范围为10°~70°,步长为0.33°;在美国麦克公司的Micromeritics ASAP 2020型吸附仪上,用氮气物理吸附测试催化剂的比表面积和孔性质,测试前先在250 ℃下脱气,用BET方程计算比表面积,用BJH方程计算孔径分布;CO化学吸附在美国麦克公司的Micromeritics ASAP 2020型吸附仪上进行,催化剂先在300 ℃下脱气10 h以去除吸附的杂质,然后在350 ℃、60 mL/min H2气流下还原6 h,在35 ℃下进行CO 吸附,并按CO/Co 化学计量比为1 来计算金属表面积;氢气程序升温还原(H2-TPR)测试在内径为6 mm 的石英反应管内进行,用QMD 质谱(OminStarTM型,德国普发真空公司)对气体信号进行在线检测,向石英管内装入50 mg样品,先在100 ℃下通入40 mL/min He气吹扫1 h,然后降温至50 ℃,将气体切成5%H2-95%He,待质谱基线平稳后,以5 ℃/min 的速率从50 ℃升温至800 ℃,并在800 ℃下保持10 min;形貌表征在日本电子株式会社JEM2100型透射电子显微镜(TEM)上进行,运行电压为200 kV;在JEM-ARM200F型球差校正的高角度环形暗场扫描透射电子显微镜(HAADF-STEM,日本电子株式会社)上观察催化剂的微观结构;用能量色散X射线分析仪(EDS,EX-230 100 m2探测器)分析元素分布;CoK边的XAS谱在台湾同步辐射研究中心(NSRRC)的BL17C1光源上进行,在催化剂进行准原位X射线吸收光谱(XAS)分析前,将催化剂压片,并用teflon膜封装,其中还原后的催化剂在不接触空气的情况下转移到手套箱内,在手套箱内进行封装,入射光能量以Co箔为标准进行校正,用气体电离探测器进行荧光信号收集,在7.5~8.5 keV 范围内扫描光子能量,前边、近边和远边的能量间隔分别为2.0,0.5和0.05 eV,所得实验数据由Athena 软件处理完成.
1.4 催化剂的活性评价
在固定床反应器上测试催化剂的费托合成反应性能,石英反应管的内径为8 mm. 称取500 mg 160~400 μm的催化剂颗粒,用2 g惰性的SiC(颗粒直径约330 μm)稀释后,装填到反应管中部,并向催化剂床层插入热电偶,监测反应过程中床层的实际温度. 在反应测试前对催化剂进行预还原,通入60 mL/min流速的氢气,在350 ℃下还原6 h,升温速率2 ℃/min. 还原后降温至220 ℃(反应温度),切成氩气吹扫30 min,然后在通入氩气的条件下,将反应体系压力升至2 MPa,待压力和温度稳定后,将Ar切换成反应气(64%H2-32%CO-4%N2),流速60 mL/min,反应24 h后,系统内的氩气被完全置换成反应气,再将反应气流速调至40 mL/min.
用3 ℃的冷阱收集低沸点的油相产物,用100 ℃的热阱收集高沸点的蜡相产物,收集到的油相和蜡相产物分别用异丙醇和二硫化碳稀释后,用带火焰离子检测器(FID)检测器的Agilent 7820A型气相色谱(GC)进行离线分析;对于未反应完全的反应气和不凝性产物,用配置FID和热导检测器(TCD)检测器的Agilent 7890B气相色谱进行在线监测,以氮气为内标作定量分析.
基于气相、油相和蜡相的结果,按下式计算碳平衡(XC):
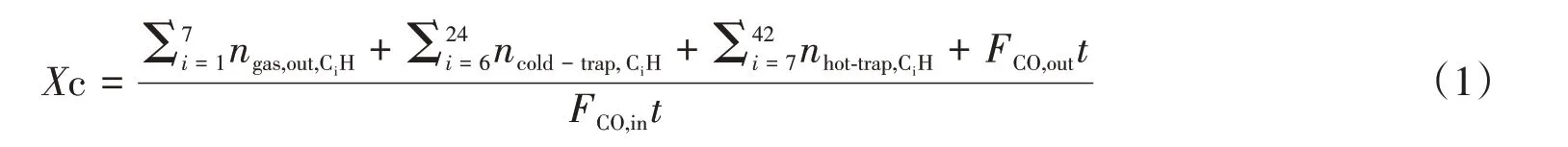
CO转化率(%)和产物选择性(SCn,%)按照下面公式计算:

式中:n表示产物包含的碳数;A表示物质在色谱中的出峰面积;AN2,blank,ACO,blank分别为反应前N2气和CO在色谱中的出峰面积;R表示物质的校正因子.
单位质量的Co在单位时间内转化CO的摩尔数(CoTY,molCO·g-1Co·s-1)按下式计算:
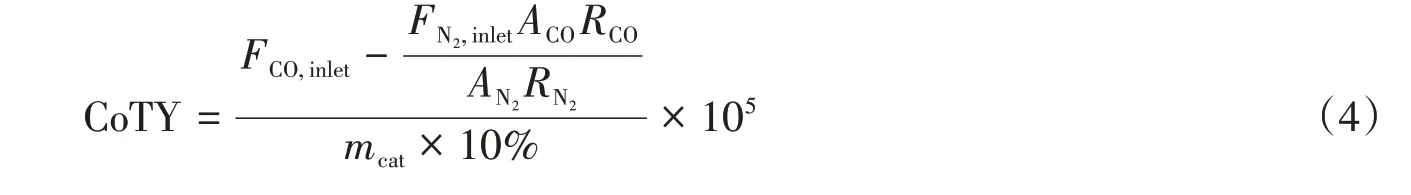
式中:FCO,inlet和FN2,inlet(mol/s)分别表示入口气体中CO和N2的流量;mcat(g)为催化剂的用量.
2 结果与讨论
2.1 催化剂的结构形貌表征
为了避免催化剂的活性金属、贵金属助剂与载体之间相互作用的影响,选取化学惰性的多孔碳化硅为载体. 多孔β-SiC作为费托合成反应的催化剂载体同时具有许多优点:导热性较高,可以减小费托合成反应过程中由于强放热而产生的温度梯度,减少热点效应;因化学惰性而具有较强的抗氧化性,与负载的金属之间相互作用弱,不会与负载金属形成难还原的化合物,有利于反应后催化剂的再生及活性组分的回收[22~24]. 载体和催化剂的氮气吸附-脱附曲线和孔径分布分别如图1(A)和(B)所示.β-SiC的氮气吸附-脱附曲线在p/p0=0.8~1.0的范围内存在回滞环,属于H3型,说明结构中存在介孔. 载体的比表面积为29 m2/g(表1).
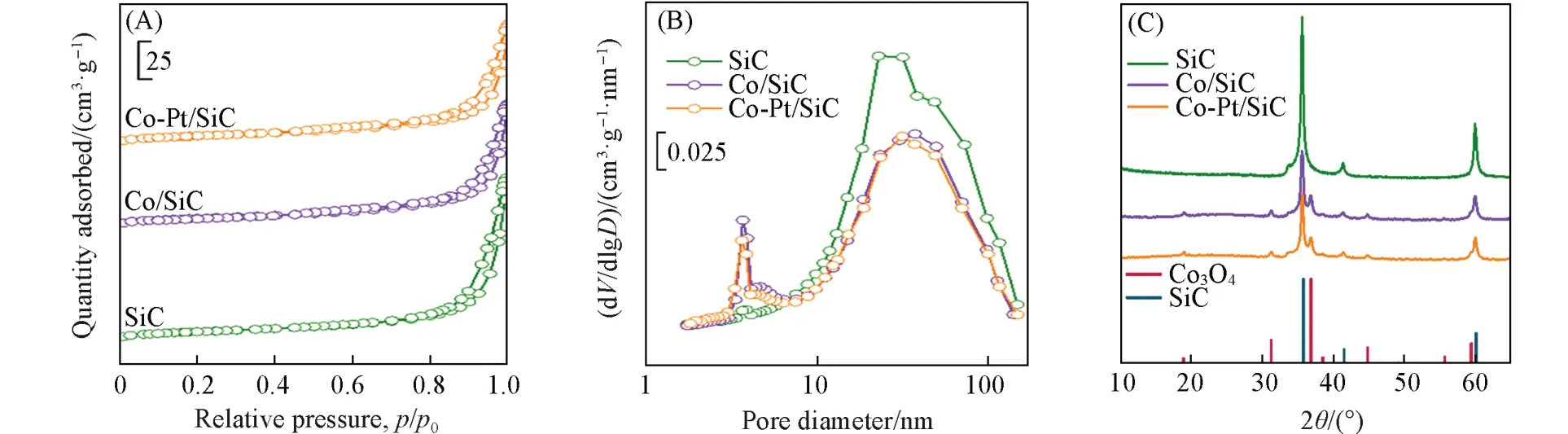
Fig.1 N2 adsorption⁃desorption isotherms(A) and pore distributions(B) of the support and catalysts and XRD patterns of the calcined catalysts and supports(C)
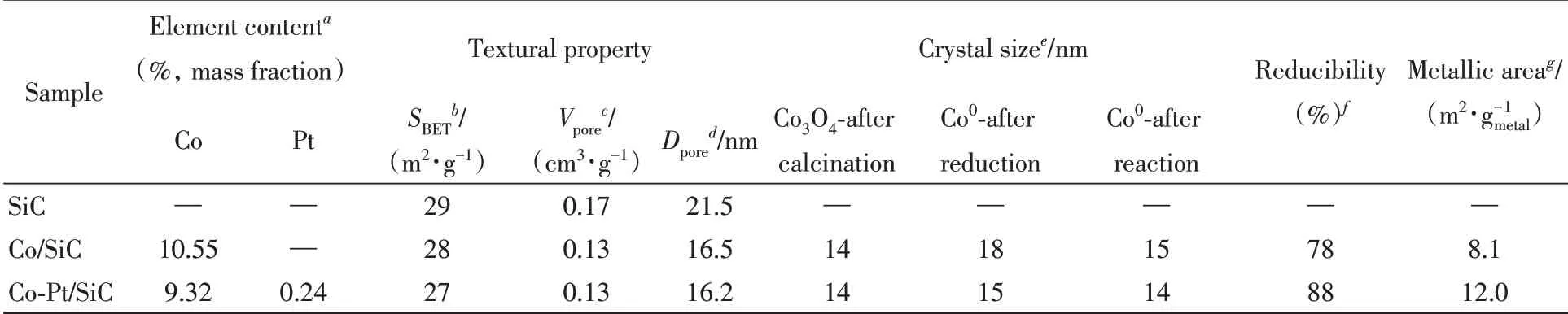
Table 1 Physical and chemical properties of the supports and catalysts
图1(B)显示,SiC的孔径主要在2~100 nm之间,即介孔和大孔,这样的多级孔结构有利于费托合成反应中反应物和产物的扩散,增加催化剂上反应物的浓度梯度,同时有利于提高长链烃类产物的选择性[25~27]. 负载金属后载体的孔结构没有发生明显变化,由于金属颗粒的分布,比表面积和孔径有所减小(表1),孔径为3 nm 的孔相对比例增多,可能是金属颗粒之间或金属颗粒和载体之间堆积形成的.
图1(C)为β-SiC 载体和焙烧后催化剂的XRD 谱图.β-SiC 的衍射峰出现在35.7°,41.4°和60.0°处,分别可归属于(111),(200)和(220)晶面. 焙烧后催化剂的XRD 谱图中,19.0°,31.3°,36.9°和44.8°处的衍射峰可分别归属于Co3O4的(111),(220),(311)和(400)晶面. 焙烧后Co/SiC和Co-Pt/SiC的衍射峰无明显差异,说明Pt的加入没有使Co3O4的晶格结构发生明显变化. XRD谱图中未见Pt物种的衍射峰,可能是因为Pt的含量低,在催化剂上呈高度分散状态.
用高分辨透射电子显微镜观察焙烧后Co3O4在SiC载体上的分散,如图2所示. 由于Co物种和SiC之间的相互作用较弱,在焙烧后的Co/SiC上,虽然发现了Co3O4的晶格[图2(C)和(F)],但没有观察到清晰的Co3O4颗粒[图2(A)和(B)]. 而对于Co-Pt/SiC催化剂,在载体的边缘处还观察到了许多分散的、直径为5~10 nm的Co3O4颗粒[图2(D)和(E)]. 因为Pt助剂的存在会促进焙烧过程中硝酸钴前驱体的分解[28,29],使氧化钴的结晶位点增多,形成的氧化钴颗粒更小,从而使得钴物种的分散度提高[30,31].

Fig.2 TEM images of calcined Co/SiC(A, B) and Co⁃Pt/SiC catalysts(D, E) and the corresponding FFT images of Co/SiC(C)and Co⁃Pt/SiC(F)
2.2 还原后催化剂的物化性质
除了促进钴物种的分散,催化剂中加入贵金属Pt 等助剂还可以显著促进氧化钴物种的还原[31].贵金属对于还原过程的促进作用与其解离氢气的能力强有关系,这个促进过程可能遵循氢溢流机理:贵金属(如Pt)解离氢气后,解离的氢物种溢流到氧化钴上,从而加速还原过程的发生[32~34]. 对催化剂进行了氢气程序升温还原(H2-TPR)测试(图3),对于Co/SiC,Co3O4的还原峰在200~400 ℃之间,其中285 ℃处的还原峰可归属于Co3O4→CoO的还原,347 ℃处的还原峰可归属于CoO→Co0的还 原. 与Co/SiC 相 比,Co-Pt/SiC 中 的Co3O4可 在350 ℃下还原完成. 此外,该样品在145 ℃下出现了一个新的还原峰,由于在50~450 ℃下没有发生Pt物种的还原,因此可以推测加入的Pt使Co3O4在较低温度下即发生了还原. 此外,计算发现加入Pt后,催化剂的还原度从78%提高到88%(表1),这可能也和Pt 助剂促进了小粒径的氧化钴颗粒的还原有关[28,32].
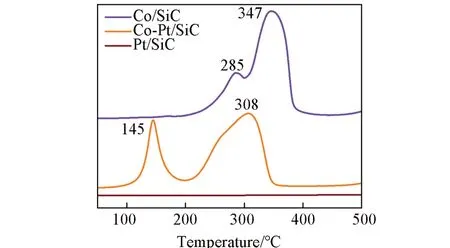
Fig.3 H2⁃TPR profiles of the catalysts
为了进一步分析还原后钴物种的精细结构,对催化剂进行了XAS表征. 图4(A)为Co的K边归一化X 射线近边吸收光谱(XANES). 对于还原后的样品,Co/SiC 和Co-Pt/SiC 的吸收边位置位于Co3O4和Co之间,说明样品中存在部分氧化钴;7712 eV处的峰对应于Co原子的1s→3d电子跃迁,Co-Pt/SiC在该处的峰强度更大,说明其金属性更强[17],这与H2-TPR的结果一致. 对应的扩展X射线吸收精细光谱(EXAFS)如图4(B)所示. 对比催化剂R空间的数据,发现焙烧后的样品第一壳层主要是Co—O键,第二壳层和第三壳层存在Co—Co键,与Co3O4标样一致,说明焙烧后的Co物种主要以Co3O4的形式存在.还原后的样品Co—O配位壳层减弱,金属钴的Co—Co配位壳层增加,主要以Co—Co键的形式存在,与Co-foil一致,说明大部分钴物种向金属态转变[26,35].
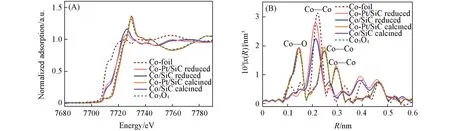
Fig.4 Quasi in situ Co K⁃edge XANES spectra(A), Fourier transform of the Co K⁃edge EXAFS(B) of the reduced catalysts,as well as reference samples
通常认为金属钴的表面是费托反应的活性位点,CO,H2的活化解离和C—C偶联过程都在金属钴表面发生[1,2,36],因此,催化剂上金属钴的性质以及表面活性钴位点的密度会直接影响其催化反应的性能. 对还原后的催化剂进行了XRD测试(图5). 经过还原后,Co3O4的衍射峰消失,在44.2°处出现了尖锐的衍射峰,归属于fcc Co的(111)晶面,此外,在47.6°处出现一个弱衍射峰,归属于hcp Co的(101)晶面,说明还原后催化剂上的钴物种主要是fcc相的金属钴,还有少量的hcp Co. 根据谢乐公式计算得到,Co/SiC和Co-Pt/SiC上fcc Co的晶粒尺寸分别为18和15 nm.
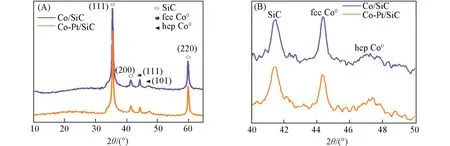
Fig.5 XRD patterns of the reduced catalysts
催化剂表面金属钴位点的密度是决定费托反应活性的关键因素,可以通过CO化学吸附对暴露出来的金属钴比表面积进行测定[21]. 根据CO 化学吸附的结果,加入Pt 后催化剂的金属表面积从8.1 m2/gCo提高到12.0 m2/gCo(表1),提高了49%. 这一方面是因为Pt促进了钴前驱体的分解在催化剂上形成了粒径较小的钴纳米粒子,另一方面也因为Pt促进了Co3O4的还原,提高了钴物种的还原度. 以上结果表明,Pt助剂从促进钴的分散和还原两个方面,使得表面金属钴数量增加,有利于费托反应活性的提高.
2.3 Pt在催化剂上的微观结构
为了更好地理解Pt 对Co/SiC 的促进作用,借助HAADF-STEM 对催化剂上Pt 的分布进行了解析.从催化剂的HAADF-STEM 照片观察到,经过还原、钝化后,Co纳米粒子呈核-壳结构[图6(A)],内核的晶格间距为0.20 nm,归属于fcc 相的Co 的(101)面;壳层的晶格尺寸为0.24 nm,是Co3O4的(311)面,氧化层的厚度为1~2 nm[图6(B)~(D)]. 从EDS 元素分析的结果中可以看出,Pt在Co纳米粒子上较均匀地分散[图6(E)~(H)].

Fig.6 High⁃angle annular dark⁃field scanning transmission electron microscopy(HAADF⁃STEM)images(A) and the corresponding EDS elemental mapping of reduced Co⁃Pt/SiC passivated under atmosphere(E—H)(B)Magnified HAADF-STEM images of Co-Pt/SiC in the frame of(A);(C)corresponding local FFTs of(A)are displayed;crystal planes assigned to Co(red circles),the crystal plane indexed to Co3O4(green circles);(D)superimposed IFFTs formed using spots in the corresponding FFT images shown in(C)[Co(red),Co3O4(green)].
从图7可以清晰地看到,Pt在Co纳米粒子上以原子级分散的形式存在:元素的原子序数越大,其在HAADF像中的强度越大,图7(A)中强度更明显的亮点为分散的Pt原子[图7(B)和(C)]. 还观察到Pt更倾向于分布在Co0上,而不是Co3O4的氧化层上. Davis等利用EXAFS发现,Pt倾向于以孤立原子的形式分散在Co上,并和Co发生紧密作用,即便是Pt的质量分数高达5%(Co质量分数为25%)时,也只检测到了Pt—Co键,而没有发现Pt—Pt键[21,37];而且在氧化-还原的循环过程中,Pt助剂的分布都和Co邻近[38]. 本文结果和Davis等的结论一致.
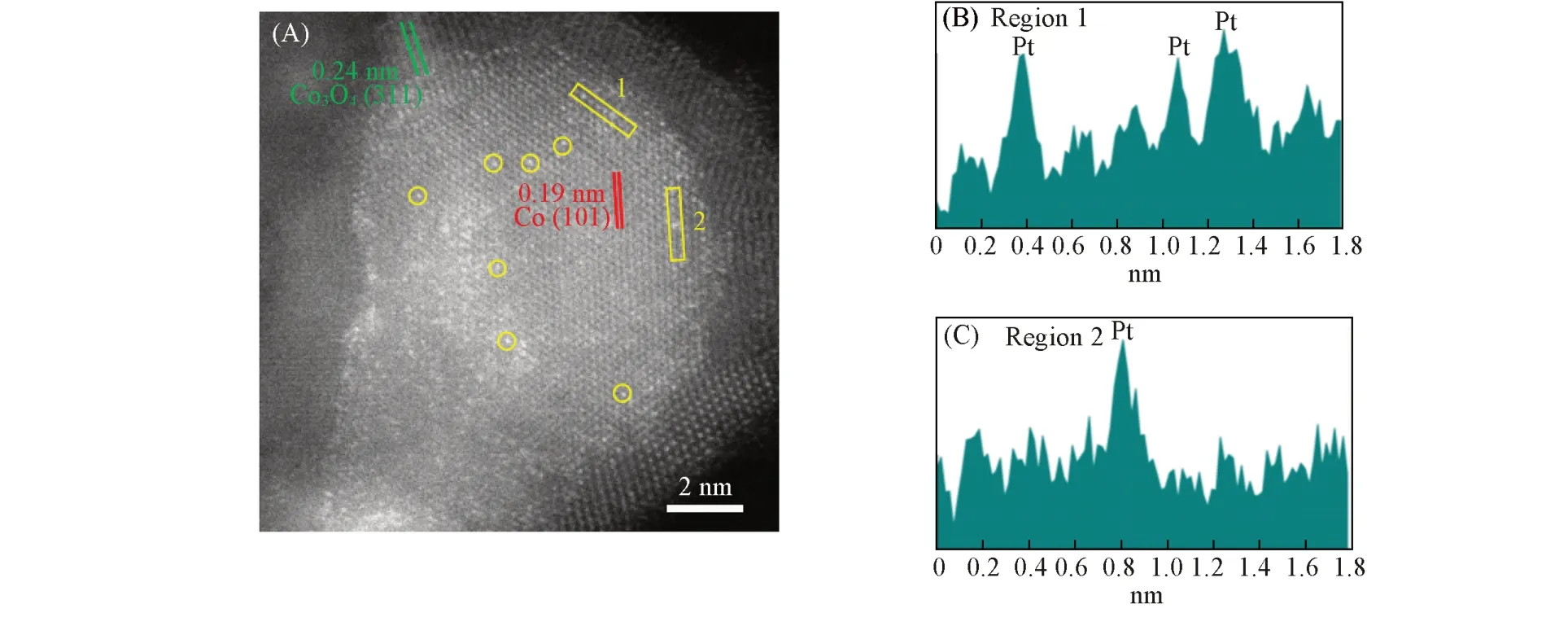
Fig.7 HAADF⁃STEM image of reduced Co⁃Pt/SiC passivated under atmosphere(A)and the corresponding intensity profiles of the selected regions in the yellow boxes in(A)(B,C)(A)Pt atoms are indicated by yellow circle.
Pt在Co上的高度分散以及和Co紧密的相互作用与其助剂效应密切相关,这样的结构有利于Co物种对于Pt上解离的氢气的利用. 在还原过程中,Pt的存在使得H2的解离可以在更低温度下发生,解离的氢物种溢流到氧化钴上,氧化钴的还原开始发生,加上金属钴自身的还原性,整个氧化钴颗粒的还原得到促进. 在反应过程中,Pt有利于促进反应物H2的解离,由此也会促进金属钴上发生的CO氢助解离[6],使得催化剂的费托合成反应活性提高.
2.4 催化剂的费托合成反应性能
对催化剂进行了费托合成反应测试来验证Pt 的促进作用,结果如图8 所示. 在70 h 的反应测试中,催化剂的转化率基本保持稳定,其中Co/SiC的CO转化率为20%,加入Pt后,CO转化率提高到31%[图8(A)],单位质量的钴上Co 的转化率(CoTY)也从3.42×10-5molCO·g-1Co·s-1提高到6.52×10-5molCO·g-1Co·s-1[图8(B)],说明加入Pt后,催化剂的费托反应活性明显提高. 转化频率(TOF)从0.17 s-1提高到0.22 s-1[图8(D)],说明Pt助剂提高了费托反应速率. 这可能是因为Pt助剂的加入促进了CO解离和加氢过程. 在费托反应中,CO 的解离和加氢引起的链终止反应被认为是影响反应速率的关键过程[36,39]. 在fcc Co上CO更倾向于发生氢助解离[6,40],即CO先经过加氢形成—COH,—HCO,—H2CO等中间体,然后才发生C—O键的断裂,该路径的能垒比CO直接解离的能垒更低[6]. 因此,催化剂上钴的加氢能力对费托反应速率有较大程度的影响. Pt助剂的加入提高了钴基催化剂解离氢气的能力,因为与钴纳米颗粒在结构上的紧密相互作用,Pt对金属钴上发生的CO氢助解离和加氢过程的促进作用也更容易发生,因此,Pt的加入可以使钴基催化剂的费托反应活性得到提高. 加入Pt后,催化剂的C5+选择性无明显变化,但C2~C5产物的烯烃/烷烃比例从1.0变为0.7[图8(C)],这是因为加入Pt后,催化剂解离氢气的能力提高,催化剂表面H/C比例增大,催化剂的加氢能力增强,使得产物中饱和加氢产物增多[33].
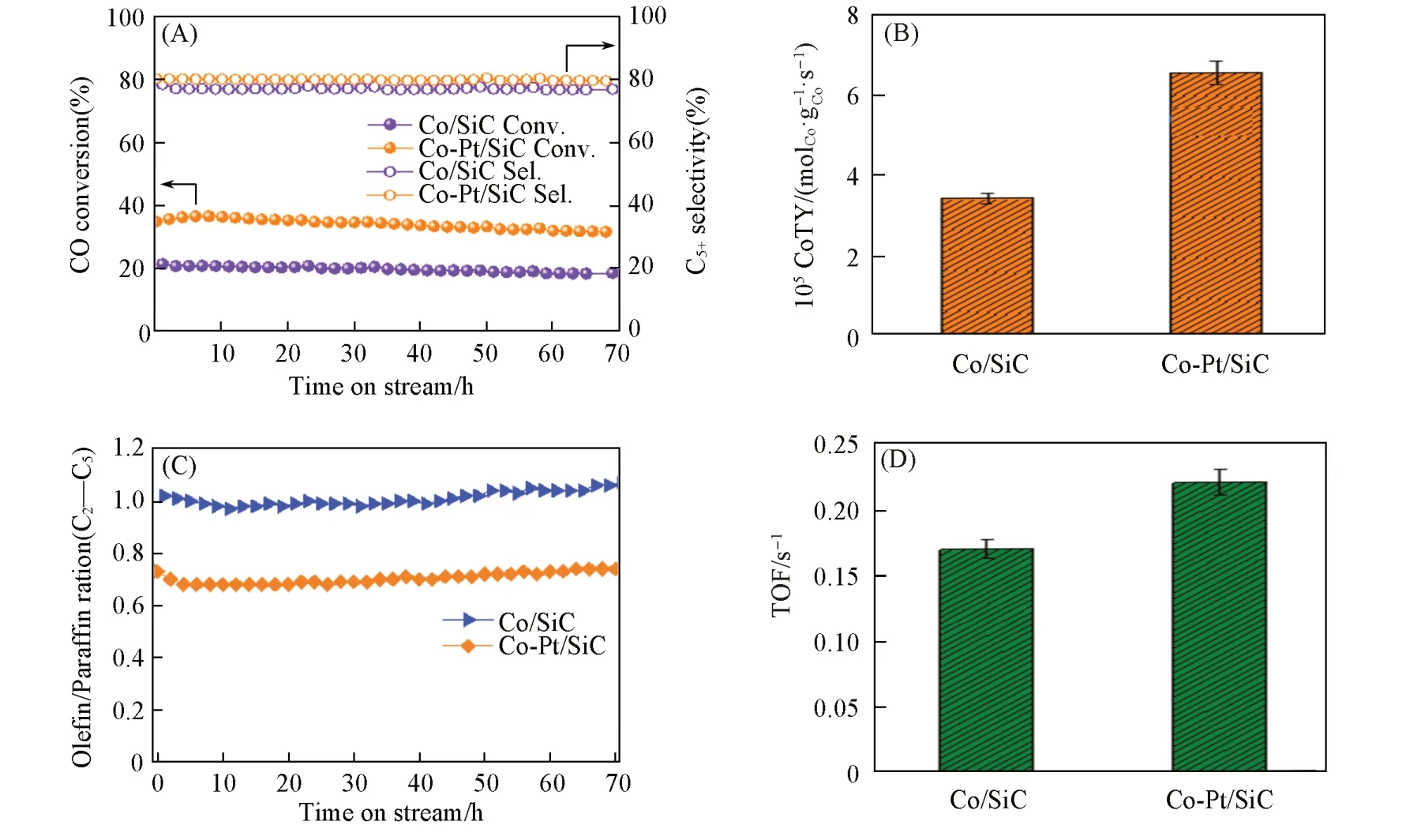
Fig.8 FTS catalytic performance of Co/SiC and Co⁃Pt/SiC catalysts(A)CO conversion as a function of time on stream;(B)CoTY of the catalysts;(C)C2—C5 olefin/paraffin molar ratio as a function of time on stream;(D)TOF of the catalysts. Reaction conditions:nH2/nCO=2,reaction temperature=220 ℃,pressure=2 MPa,GHSV=4800 mL·h-1·g-ca1t.
对反应后的催化剂进行了XRD测试,结果如图9(A)和(B)所示,在21.8°和24.2°处出现了较强的衍射峰,为富集在催化剂表面的烃类产物. 载体SiC的衍射峰无明显变化;在44.2°处出现的尖锐衍射峰为fcc Co(111)面;在47.6°处观察到hcp Co(101)面的衍射峰,说明催化剂上少量的hcp Co能在反应过程中稳定存在. 根据谢乐公式,反应后Co/SiC 和Co-Pt/SiC 上fcc Co 的晶粒尺寸分别为15 和14 nm,与反应前的基本相同(分别为18和15 nm),说明在反应过程中没有发生明显的金属烧结,Co0的晶相结构基本保持稳定.

Fig.9 XRD patterns of the used catalysts Co/SiC(A)and Co⁃Pt/SiC(B)
3 结论
选取高导热导电性、化学惰性的多孔碳化硅为载体,考察了Pt对Co基费托催化剂的促进作用. 结果表明,Pt的加入一方面增加了焙烧过程中氧化钴的结晶位点,促进了氧化钴的分散;另一方面降低了氧化钴的还原温度,提高了还原度,最终使得Co0表面积增大,费托合成反应的活性相增多,有利于催化活性的提高. 利用原子分辨的HAADF-STEM观察到,Pt以单分散形式分布在Co纳米粒子表面,这样的结构特点有利于Pt解离的氢溢流到Co物种表面,既有利于促进氧化钴的还原,使Co0活性位点增多,也有利于促进费托反应中H2的活化解离以及CO的氢助解离,使得反应速率提高.
感谢日立公司Hiroaki Matsumoto 先生在球差电镜测试过程中提供的支持. 感谢NSRRC 实验室SU Bingjian博士在同步辐射测试中的帮助.
猜你喜欢
华人时刊(2022年9期)2022-09-06
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
华人时刊(2020年15期)2020-12-14
今日农业(2020年20期)2020-11-26
中国洗涤用品工业(2015年9期)2015-02-28
中国洗涤用品工业(2015年5期)2015-02-28
中国火炬(2013年11期)2013-07-25
中国火炬(2013年10期)2013-07-24
