
愈创木基丙三醇-β-愈创木基醚对土壤中多环芳烃的归趋的影响及微生物群落效应的研究
2022-09-21 03:34季聪杰安雪晖陈禹竹
安徽农业科学 2022年17期
尹 晗,季聪杰,安雪晖,孙 月,陈禹竹*
(1.中电建路桥集团有限公司,北京 100048;2.清华大学水利水电工程系,北京 100084;3.中国科学院南京土壤研究所,江苏南京 210008)
多环芳烃(PAHs)是含有2个或2个以上苯环连接在一起的烃类化合物,随着其环数增加、化学结构的变化和疏水性的增强,其化学稳定性、持久性、抗生物降解能力和毒性都会增强,多环芳烃已成为我国土壤中主要的有机污染物之一,被认定为影响人类健康的主要有机污染物。一些高分子量有机物不能直接作为大部分微生物的唯一碳源和能源,在有其他化合物作为一级底物提供碳源和能源时,该有机物才能作为二级底物被降解,这种现象称为共代谢作用,这类一级底物被称为共代谢底物。多环芳烃是一类难降解的有机污染物,分子量高,很少有微生物可以直接利用其作为唯一碳源,因此,高分子量多环芳烃通常以共代谢方式进行降解,共代谢过程已作为一种生物技术在芳香族化合物生物降解修复中得到广泛应用。
木质素是一类复杂的有机聚合物,其化学结构与多环芳烃结构类似,已有研究表明,木质素具有刺激微生物降解芳香族污染物的作用。木质素解聚产生大量双芳基或单芳基降解产物,它们的进一步代谢会诱导微生物产生胞外酶等物质,这些可能会促进多环芳烃降解。木质素促进芳香族污染物的降解过程,可能涉及共代谢机制,但其具体的微生物共代谢机制尚不明确。木质素在土壤中的降解过程涉及多种微生物,降解的不同阶段也具有不同的共代谢效应。目前解决多环芳烃的污染问题以及深入地研究木质素促进多环芳烃降解的微生物共代谢机制对研究多环芳烃污染土壤的修复具有重要意义。因此,该研究选择了一种木质素模型二聚体化合物愈创木基丙三醇-β-愈创木基醚(guaiacylglycerol-β-guaiacyl ether,GGE)为刺激底物,以多环芳烃苯并[a]蒽(Benz(a)anthracene,BaA)为模式污染物,同时结合真菌抑制剂放线菌酮(cycloheximide),设立微宇宙培养试验,结合C同位素示踪技术、定量PCR技术和扩增子测序来分析GGE对BaA的归趋的影响及其微生物群落效应。
1 材料与方法
土壤采集于南京市郊农田,自然风干,研磨后过2 mm网筛,保存于4 ℃,用于后续培养试验。土壤理化性质:pH 6.84、总碳12.7 g/kg、总氮1.3 g/kg、全磷0.57 g/kg、全钾19.3 g/kg、田间持水量33.3%。
[7,12-C]苯并[a]蒽(C-BaA,10 mCi/mmol,99%纯度),美国Radiolabeled Chemicals公司;苯并[a]蒽(C-BaA,98%纯度),梯希爱(上海)化成工业发展有限公司;真菌抑制剂放线菌酮,西格玛奥德里奇(上海)贸易有限公司。液体闪烁分析仪(LSC),美国贝克曼库尔特有限公司;生物氧化燃烧仪(OX-500),德国Zinsser Analytic公司。
设立2组微宇宙培养试验,2组处理相同,微宇宙培养试验在60 mL矿化管中进行,每管加入5.0 g土壤样品,土壤持水量为60%,处理组添加GGE的含量为500 μg/g,真菌抑制剂放线菌酮的含量为1 mg/g。
第一组通过同位素示踪技术,检测GGE处理组和对照组在有无放线菌酮存在的情况下C-BaA的环境归趋,每个处理设3个平行。添加C标记的BaA至土壤样品中,使其终浓度为50 μg/g,放射性为2.0×10DPM。在矿化管内放置装有1 mL 1 mol/L NaOH溶液的小瓶子,收集释放的CO,培养时间为70 d,每7 d取样测定培养过程中释放的CO量,利用液体闪烁分析仪(LSC)测定NaOH溶液吸收的CO含量。
培养结束后,将矿化管内的土壤样品风干,过2 mm网筛,取2.0 g用二氯甲烷在50 ℃下索氏提取24 h,将萃取物旋蒸后用环己烷熔解,经LSC测定可提取态(DCM)含量。不可提取态(NER)进一步分离为富里酸(FA)、腐殖酸(HA)和胡敏素(HU)结合态。参照Shan等的方法,将DCM提取后的土样风干,用0.1 mol/L无氧NaOH在250 r/min下振荡提取24 h,12 000 r/min离心30 min,所得沉淀为胡敏素,上清液为富里酸和腐殖酸的混合溶液,在上清液中加入6 mol/L HCl调节混合溶液pH至1,4 ℃下沉淀24 h,7 000 r/min离心30 min,上清液为FA组分,利用LSC检测FA和HA组分的放射性,得到FA、HA结合态的含量。将胡敏素沉淀冷冻干燥后,用生物氧化燃烧仪在900 ℃下完全氧化4 min,收集氧化后产生的C标记的CO,并用LSC检测HU组分放射性,得到HU结合态的含量。
第二组添加未标记的C-BaA至土壤样品中,使其终浓度为50 μg/g,用于检测培养过程中的微生物变化,每个处理6个重复,分别在培养中期(第42天)和培养结束(第70天)时,各取3支破坏性取样。土壤样品置于-20 ℃保存,用于后续微生物分析。使用FastDNA SPIN Kit for Soils土壤DNA提取试剂盒,参照试剂盒说明书操作,提取土壤样品中的DNA。测定DNA浓度后,置于-20 ℃保存。
采用定量PCR的方法,测定各样品中细菌16S rRNA基因、18S rRNA基因、甲醇脱氢酶基因和革兰氏阳性菌多环芳烃双加氧酶GP PAH-RHDα 基因表达丰度。土壤样品总DNA稀释至10浓度,作为定量模板,定量PCR反应体系 (20 μL):Top Green qPCR SuperMix (SYBR Green) 10.0 μL,正、反向引物 (10 μmol/L)各0.4 μL,ddHO 7.2 μL,模板DNA 2.0 μL。采用细菌16S rRNA基因序列PCR通用引物Eub338F (5′-ACTCCTACGGGAGGCAGCAG-3′)和Eub518R (5′-ATTACCGCGGCTGCTGG-3′),扩增目的片段;定量PCR扩增程序:95 ℃ 3 min;95 ℃ 30 s,56 ℃ 30 s,72 ℃ 30 s,35 个循环;72 ℃ 10 min。采用真菌18S rRNA基因序列PCR通用引物nu-SSU-0817 (5′-TTAGCATGGAATAATRRAATAGGA-3′)和nu-SSU-1536 (5′-ATTGCAATGCYCTATCCCCA-3′),扩增目的片段;定量PCR扩增程序:94 ℃ 3 min;94 ℃ 30 s,53 ℃ 30 s,72 ℃ 30 s,35 个循环;72 ℃ 10 min。采用基因序列PCR通用引物mdh1 (5′-GCGGIWSCAICTGGGGYT -3′)、 mdh2 (5′-GCGGIWSGAICTGGGGYT -3′)和mdhR (5′-GAASGGYTCSYARTCCATGCA -3′),扩增目的片段;定量PCR扩增程序:95 ℃ 5 min;95 ℃ 1 min,53 ℃ 1 min,72 ℃ 1 min,40 个循环;72 ℃ 10 min。采用GP PAH-RHDα基因序列PCR通用引物GP RHDα F (5′-CGGCGCCGACAAYTTYGTNGG -3′)和GP RHDα R (5′-GGGGAACACGGTGCCRTGDATRAA-3′),扩增目的片段;定量PCR扩增程序:95 ℃ 3 min;95 ℃ 20 s,54 ℃ 30 s,72 ℃ 30 s,40 个循环;72 ℃ 10 min。
利用Illumina Miseq高通量测序技术,对细菌16S rRNA基因和真菌ITS基因测序,分析土壤细菌和真菌群落组成。测序工作送至上海美吉生物医药科技有限公司完成。所得原始数据使用QIIME (V1.9.1)和USEARCH (V8.1.1861)流程进行分析,序列经拼接、过滤和质控后,在97%相似性水平划分操作分类单元(operational taxonomic unit,OTU),细菌序列通过与SILVA数据库(https://www.arb-silva.de)比对确定系统学分类,真菌序列通过与Unite数据库(http://unite.ut.ee/)比对确定系统学分类,得到每个样品的OTUs和物种注释的基本分析结果,同时对物种注释在各个分类水平上进行组成结构的统计分析。再对OTUs进行丰度、多样性指数等分析。
利用SPSS 20.0、 Excel 2019和Graphpad Prism 8对数据进行整理分析与作图,利用Galaxy平台(http://huttenhower.sph.harvard.edu/galaxy/)进行LefSe分析。
云工作流引擎的设计目标主要是将工作流引擎从应用系统中解耦出来,形成一个独立的服务,从而降低系统的耦合性,提供更加灵活的服务支持,实现复杂度可控、独立部署、技术选型灵活、服务容错和按需独立扩展等目标。
2 结果与分析
通过同位素示踪技术,测定C-BaA在土壤中的归趋情况,如图1所示。在培养的前期阶段,矿化率呈现CK>GGE>CK-cycloheximide>GGE-cycloheximide的趋势,在70 d的培养结束后,GGE的矿化率最高,达到4.61%,CK的矿化率为4.43%,略低于GGE。除矿化部分外,大部分的C-BaA可被二氯甲烷提取,CK组和GGE处理组中的可提取态C-BaA分别为71.17%和70.23%。不可提取态进一步细分为富里酸(FA)、腐殖酸(HA)和胡敏素(HU)结合态,在CK组中,与富里酸、腐殖酸和胡敏素结合的比例分别为3.37%、6.55%和14.10%,在GGE处理组中,与富里酸、腐殖酸和胡敏素结合的比例分别为3.13%、6.78%和14.75%。
在CK组和GGE处理组中,真菌抑制剂放线菌酮(cycloheximide)的添加对BaA在土壤中的归趋有不同程度的影响。CK组受到放线菌酮的影响较小,矿化基本未受影响,除腐殖酸结合态减少了13.13%,其他组分变化很小。在GGE处理组中,加入放线菌酮后,矿化率减少,仅为3.50%,矿化率降低了22.35%,富里酸结合态增加了29.71%,胡敏素结合态增加了15.39%。
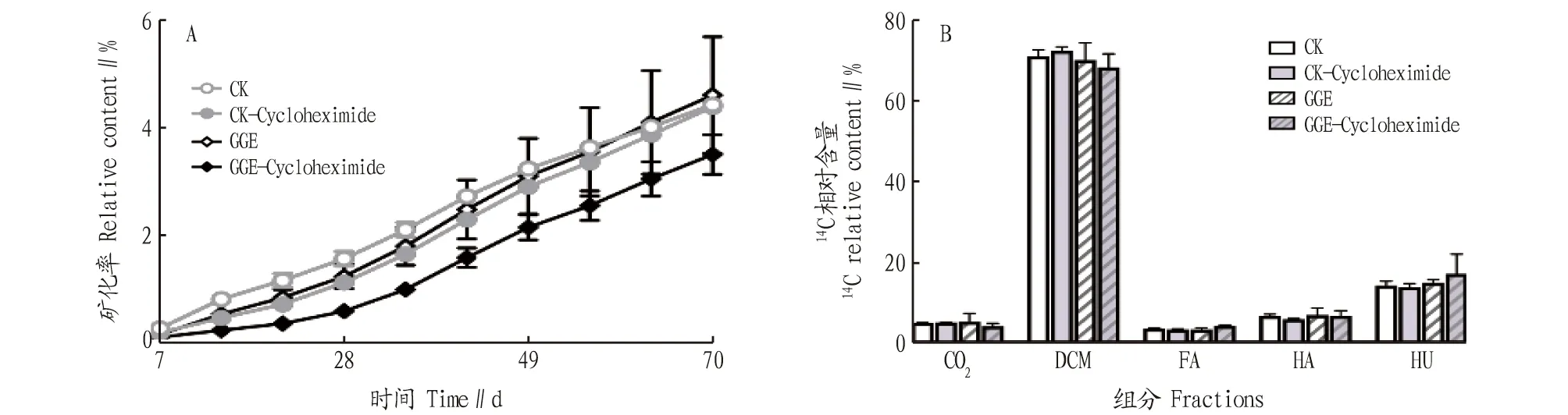
图1 培养过程中的14CO2矿化情况(A)以及培养结束时14C-BaA的归趋(B) Fig.1 The mineralization of 14CO2 during the incubation(A) and the distribution of 14C-BaA at the end of incubation (B)
采用定量PCR的方法,检测了土壤中细菌16S rRNA基因、18S rRNA基因和革兰氏阳性细菌多环芳烃双加氧酶GP PAH-RHDα基因,结果如图2所示。不添加放线菌酮的情况下,细菌16S rRNA基因中,在CK组和GGE处理组的培养中期的基因拷贝数分别为1.02×10和8.89×10copies/g,培养结束后的基因拷贝数分别为3.57×10和3.92×10copies/g,CK和GGE处理之间未见显著差异;真菌18S rRNA基因中,GGE处理组相对于CK组提高了基因拷贝数,从1.92×10copies/g提高至2.70×10copies/g,培养结束后,拷贝数均明显下降,且处理间相差不大;与CK组相比,GGE处理组也提高了甲醇脱氢酶的基因拷贝数,在培养期间从1.09×10copies/g提高至1.79×10copies/g,培养结束后整体上拷贝数减少;在GP PAH-RHDα基因中,随着培养的进行,CK组的基因拷贝数逐渐减少,从1.78×10copies/g减少至1.17×10copies/g,而GGE处理组的拷贝数逐渐增加,从1.33×10copies/g增加至1.75×10copies/g。放线菌酮添加的情况下,整体上没有明显减少土壤中细菌16S rRNA基因、真菌18S rRNA基因和基因的数量,但在GP PAH-RHDα基因中,放线菌酮的添加显著抑制了GGE处理组中基因丰度。
对细菌16S rRNA基因的高通量测序结果进行分析,一共获得了55万条序列,经质量控制,单个样品的序列数在15 978~34 374,在97%序列相似性水平上获得24 750个OTU。对真菌ITS基因的高通量测序结果进行分析,一共获得了78万条序列,经质量控制,单个样品的序列数在24 393~71 441,在97%序列相似性水平上获得1 579个OTU。
对相对丰度>1%的细菌门/纲类进行分析,结果发现(图3A),在第42天,CK组中主要为α-变形菌纲(Alpha-proteobacteria)、β-变形菌纲(Beta-proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和γ-变形菌纲(Gamma-proteobacteria),占比分别为14.47%、17.07%、21.47%、12.14%、5.93%和4.97%。GGE处理组增加了α-变形菌门、拟杆菌门(Bacteroidetes)的相对丰度,而β-变形菌纲和芽单胞菌门(Gemmatimonadetes)则有所减少。各类细菌门/纲的相对丰度在培养时间上也存在着一定的变化,第70天与第42天相比,CK和GGE处理下酸杆菌门和α-变形菌纲的相对丰度明显增加,β-变形菌纲、放线菌门和厚壁菌门的比例均有减少。
对真菌在门水平上进行分析,结果发现(图3B),在第42天时,CK组中主要真菌为子囊菌门(Ascomycota)、担子菌门(Basidiomycota)和壶菌门(Chytridiomycota),占比分别为63.67%、23.60%和2.28%,GGE处理组与CK组相比,子囊菌门的相对丰度增加至73.85%,担子菌门的相对丰度减少,为11.58%。各类真菌的相对丰度在培养时间上也存在着一定的变化,到第70天时,CK和GGE处理中的子囊菌门的相对丰度有所减少,CK组与第42天相比,被孢霉门(Mortierellomycota)明显增加,GGE处理组中被孢霉门和球囊菌门(Glomeromycota)的相对丰度明显增加。
在OUT水平上,对细菌和真菌的群落进行非度量多维尺度(NMDS)分析(基于Bray-Curtis 距离),如图4所示。在细菌群落中,第42天和第70天的样品存在很明显的分离,表明样品在培养期间存在时间上的演替,GGE处理组的大部分样品能相对集中在一起,且与CK组之间呈现出一定的分离,表明GGE的添加对细菌群落组成产生了一定的影响。在真菌群落中,GGE处理对真菌群落影响较小,2组样品未发生分别聚类,与CK组的群落结构相似,在不同的培养时间分布上,CK组未出现明显分离,而GGE处理组呈现了一定程度的分离。
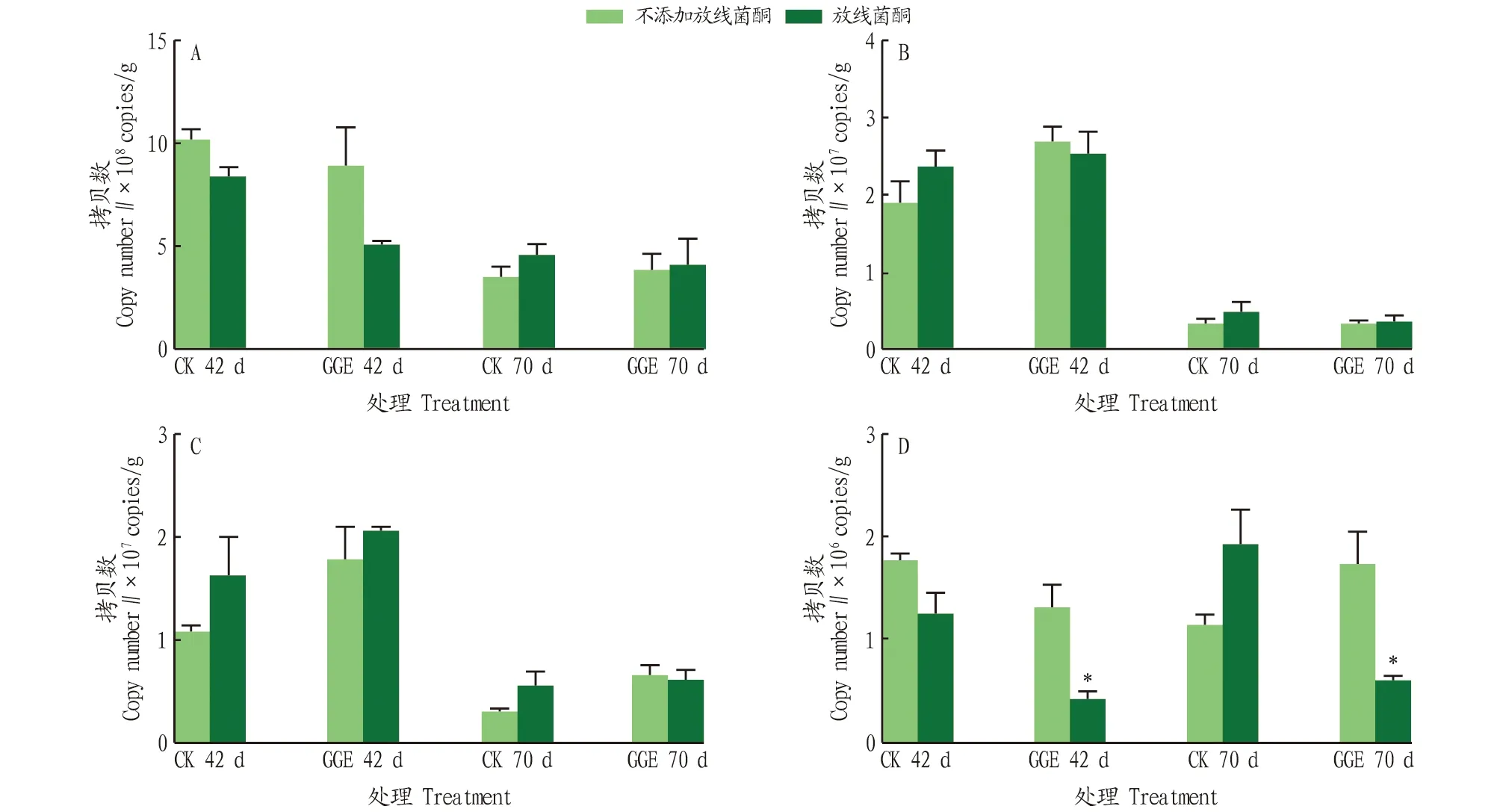
注:*表示在0.05水平上放线菌酮处理间差异显著 Note:* indicates significant difference between cycloheximide treatments at the 0.05 level图2 细菌(A)、真菌(B)、甲醇脱氢酶mxaF/xoxF(C)和 GP PAH-RHDα(D)基因的定量PCR结果Fig.2 Quantitative PCR results of bacteria(A) ,fungi (B),methanol dehydrogenase mxaF/xoxF(C) and GP PAH-RHDα genes (D)
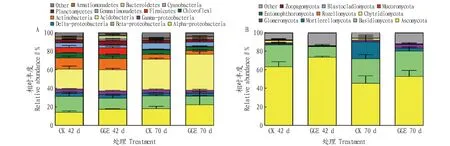
图3 细菌(A)和真菌(B)在门/纲水平上的相对丰度Fig.3 Relative abundance of bacteria (A)and fungi(B) at phylum or class level

图4 细菌 (A)和真菌(B)的非度量多维尺度(NMDS)分析Fig.4 Non-metric multidimensional scaling (NMDS) analysis of bacteria (A) and fungi (B)
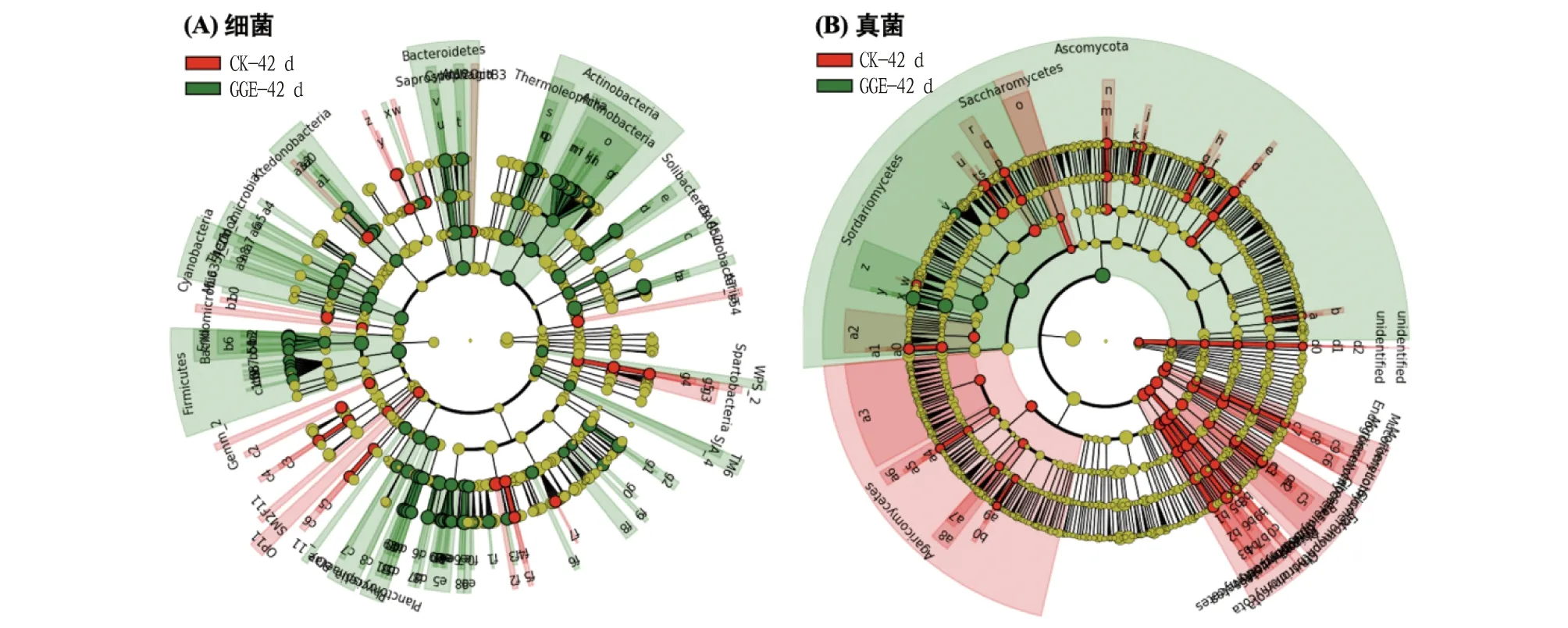
注:图中结点从内圈至外圈代表各个分类水平(界、门、纲、目、科、属),不同颜色代表在各自处理中显著富集的菌属 Note:The nodes in graphs from the inner ring to the outer ring represent the different taxonomic levels (domain,phylum,class,order,family and genus),different colors represent the significant enrichment in respective treatments图5 不同处理的微生物群落LefSe分析Fig.5 LefSe analysis of microbial communities from different treatments
3 讨论与结论
木质素是一种芳香性聚合物,被解聚后会有不同的芳基单体产生,因其与多环芳烃都含有芳香性结构,可能会成为多环芳烃降解过程中的共代谢底物,从而促进污染物的代谢。在该研究中,通过比较对照(CK)组和添加木质素的一种模式化合物的GGE处理组对土壤中BaA归趋的影响,并通过添加真菌抑制剂,来验证污染物归趋过程中细菌和真菌的贡献。从BaA的归趋结果来看,CK组与GGE处理组的矿化率相近,在CK组中加入放线菌酮对矿化几乎没有影响,而与CK组相比,GGE处理受到了放线菌酮的抑制,矿化率减少了22.35%。矿化过程主要与微生物群落有关,认为在CK组中真菌起到的作用不大,但是加入了GGE之后,真菌的作用增大,尽管矿化结果并没有显著变化,但结合微生物群落的变化,GGE的加入在一定程度上使得真菌的贡献在提升。
目前已知真菌与细菌使用完全不同的途径对多环芳烃进行转化,真菌转化多环芳烃的途径主要是胞内P450氧化酶系转化途径和胞外木质素氧化酶转化途径;而细菌对多环芳烃的转化一般属于好氧降解途径,其中双加氧酶途径是细菌降解多环芳烃最常见的方式。这些不同导致 PAHs 在转化的中间产物和转化效率等方面存在很大差异。该研究土壤微宇宙中BaA的矿化可能涉及真菌和细菌的协同作用。从定量的分析结果可以看到,在CK处理中加入放线菌酮,并未对多环芳烃双加氧酶GP PAH-RHDα基因的丰度产生明显影响,而在GGE处理中加入放线菌酮,则显著抑制了双加氧酶基因的丰度。真菌对PAHs的作用是多样的,但真菌较少能直接利用高分子量的多环芳烃,更多是以共代谢机制降解PAHs,即利用其他碳源生长的同时,在次级代谢过程中转化PAHs。培养体系中GGE的加入可能会刺激土壤中的真菌,使其先被利用,转化成某些中间体化合物,它们的进一步代谢可能会诱导土壤中的细菌表达双加氧酶等,这些酶类会作用于污染物,促进降解过程。底物基质的添加可能会引起多环芳烃的不同微生物降解机制。
根据该研究对微生物群落的分析结果,GGE的添加相比于真菌对细菌群落的影响更大。定量PCR的结果显示,在GGE处理下,基因有一定程度的增加,甲醇脱氢酶是微生物甲基代谢中的关键酶之一,LEfSe的分析结果也表明,GGE处理组显著富集了Hyphomicrobiaceae,已有研究发现为一种甲基营养菌,一些甲基营养菌已被证明可降解石油碳氢化合物和芳香族碳氢化合物,还可以利用废气中的多环芳香烃,甲基营养菌这类潜在降解菌的富集,可能也会对多环芳烃的降解有潜在能力。GGE处理组还显著富集了Rhodospirillaceae、Sinobacteraceae、Acidobacteriaceae、Burkholderiaceae等与有机物降解相关的细菌科,真菌的富集主要集中在子囊菌门粪壳菌纲。已有研究表明,子囊菌门是污染环境中的主要真菌,具有转化或去除PAHs的能力。张娟琴等从工厂附近的土壤中分离到一株可以降解PAHs的伯克氏菌(),且菌株具有较高的双加氧酶活性。Rhodospirillaceae和Acidobacteriaceae等菌科也被研究发现是PAHs的潜在降解菌。这些微生物的富集可能会在整体上提升土壤中的芳香族化合物的代谢功能,增强对多环芳烃类污染物的降解能力。
现阶段,鉴于微生物群落的复杂响应,GGE处理对土壤微生物群落的调控与多环芳烃的转化关系仍需进一步分析,希望随着研究的深入,将木质素及其代谢产物的共代谢刺激作用机制进一步的完善,为利用微生物群落降解环境污染物提供更多的科学依据。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
科学家(2021年24期)2021-04-25
上海包装(2019年8期)2019-11-11
天津造纸(2016年1期)2017-01-15
中国造纸学报(2015年1期)2015-12-16
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
中国塑料(2014年4期)2014-10-17
丝绸(2014年5期)2014-02-28
