
含有席夫碱配体的钌配合物的合成、表征及抗肿瘤活性
2022-10-10 05:43贾士芳郝秀丽温艳珍
无机化学学报 2022年10期
贾士芳 郝秀丽 温艳珍 张 燕
(太原科技大学化学与生物工程学院,太原030021)
随着顺铂类配合物作为抗肿瘤药物的发现及临床应用[1],金属配合物作为抗肿瘤药物的研究成为了一个热门课题。金属钌配合物是继铂类配合物后被认为最具有潜力的低毒高效的抗肿瘤药物[2-7]。目前部分金属钌配合物已被证实对某一种肿瘤细胞有效甚至进入临床研究阶段。1987年,Keppler等合成了对结肠癌有明显效果的[HL][trans-RuバL2Cl4](其中L为含氮杂环配体)配合物[8]。1998年,第一个进入临床研究的对转移瘤有明显抑制作用的NAMI型钌配合物由Alessio[9]合成并已进入临床二期研究。2008年,德国的Schatzschneider[10]报道合成的金属钌配合物[Ru(bpy)2(dppn)]2+通过插入模式与DNA结合后,具有与传统顺铂类药物对人乳腺癌及结肠癌细胞相近的半抑制率。2009年,Keppler等发现已经进入临床一期的钌配合物KP1019[11]对Lewis肺癌和原发性直肠癌有明显作用。
随着合成技术的发展,不断涌现出各种新型席夫碱类化合物。席夫碱类化合物可以应用到生物分析技术、金属有机配体、荧光探针[12-13]、抗肿瘤[14-19]等方面。我们以4,4′-二(4-氨基苯氧基)联苯、2,2′-二(4-氨基苯氧基)苯基丙烷、4,4′-二(4-氨基苯氧基)二苯砜为原料,分别与2-吡啶甲醛反应生成对应的席夫碱配体。原料都具有对称结构,生成的配体都是对称的双齿配体,配体与二氯二联吡啶钌以1∶2反应,生成了对应的双核金属钌配合物。
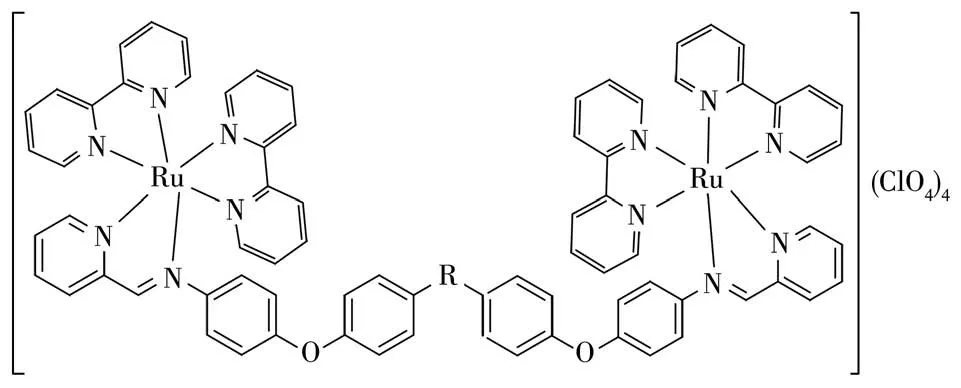
图1 含有不同桥基团的席夫碱的三种双核金属钌配合物结构图Fig.1 Structure of three dinuclear metal ruthenium complexes of Schiff bases with different bridging groups
1 实验部分
1.1 试剂与仪器
三水合三氯化钌、2,2′-联吡啶(bpy)、4,4′-二(4-氨基苯氧基)联苯、2,2′-二(4-氨基苯氧基)苯基丙烷、4,4′-二(4-氨基苯氧基)二苯砜、2-吡啶甲醛、氘代三氯甲烷、氘代二甲基亚砜等均为市售分析纯试剂,购于武汉申试化工有限公司。所有试剂都未经纯化直接使用,使用的水为蒸馏水。胎牛血清购自美国Gibco公司。
主要仪器有AL204分析天平(梅特勒-托利多仪器有限公司)、DF-101B恒温磁力搅拌器(郑州长城科工贸有限公司)、SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司)、PE20005型元素分析谱仪(美国PE公司)、Varian Mercury VX-300型核磁 共振仪(瑞士Bruker公司)、Nicolet38型傅里叶红外光谱仪(Thermo Electron Corporation)、TU-1901型紫外可见分光光度计(北京普析通用仪器有限责任公司)、Trace MS 2000GC/MS ESI质谱仪(德国NETZSCH公司)、Shimadzu RF-5301 PC荧光分光光度计(北京普析通用仪器有限责任公司)、CHI630E电化学工作站(上海辰华仪器有限公司)、EPICS XL-MCL型酶标仪(美国Beckman Coulter公司)、Varioskan Flash型多功能酶标仪(美国Thermo Scientific公司)。
1.2 目标配合物的合成
1.2.1 合成路线图
配体及对应配合物的具体合成路线如图2所示。
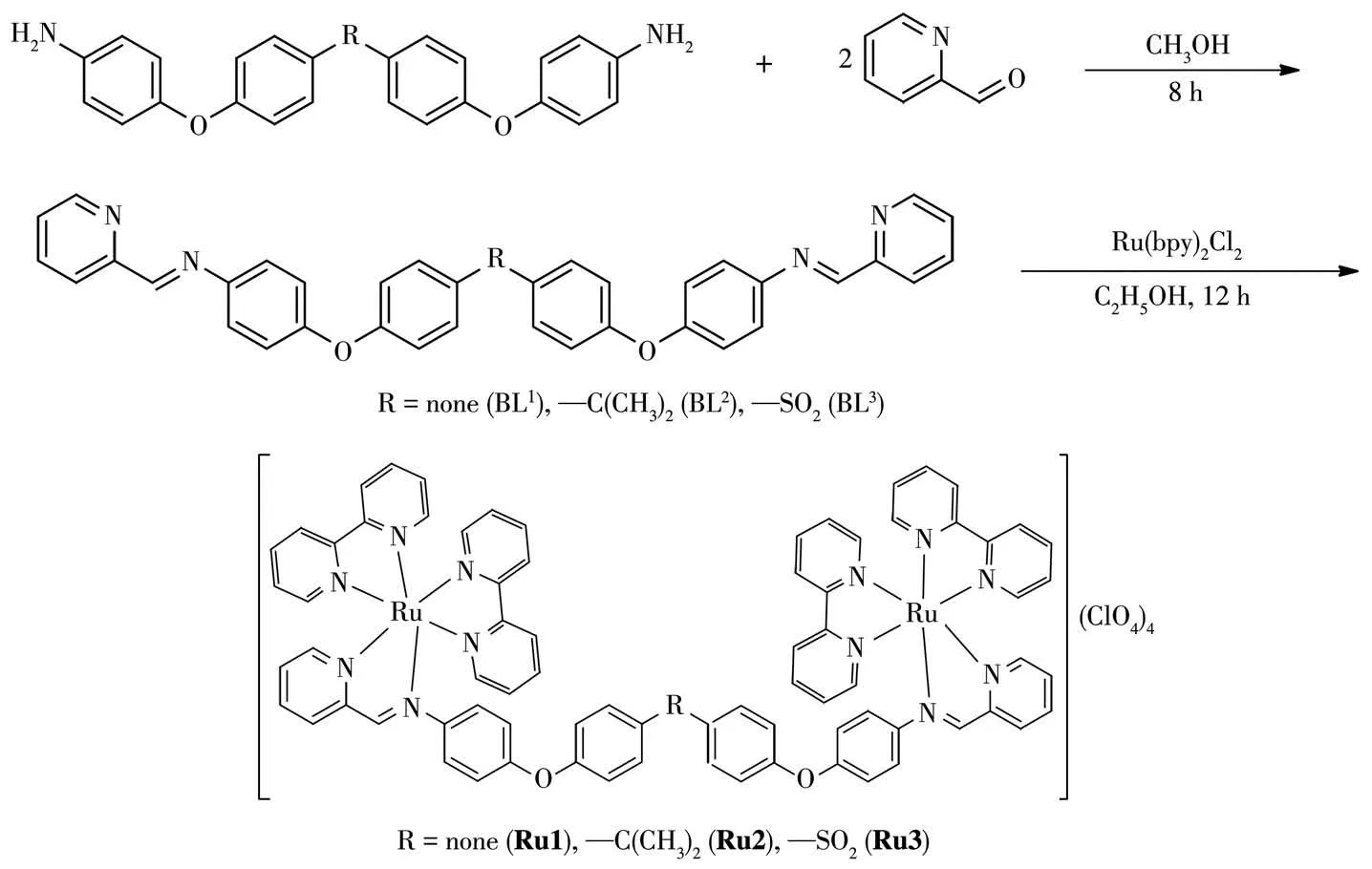
图2 席夫碱配体BL1~BL3及对应的配合物Ru1~Ru3的合成路线Fig.2 Synthesis of Schiff base ligands BL1-BL3 and complexes Ru1-Ru3
1.2.2 配体及配合物的合成
配体BL1的合成:称取0.37 g(1 mmol)4,4′-二(4-氨基苯氧基)联苯和0.54 g(5 mmol)2-吡啶甲醛放入100 mL的圆底烧瓶中,加入35 mL甲醇,加热搅拌回流8 h,有黄色固体产生,过滤得到黄色固体,用甲醇和蒸馏水各洗3次,真空干燥,得到0.48 g配体BL1,产率为88%。元素分析(C36H26N4O2)计算值(%):C,79.1;H,4.8;N,10.3。实验值(%):C,79.2;H,4.8;N,10.4。1H NMR(CDCl3):δ8.80(2H,d,J=3.0 Hz,Py),8.64(2H,s,CH),8.19(2H,d,J=6.0 Hz,Py),7.81(2H,t,J=9.0 Hz,Py),7.54(4H,d,J=6.0 Hz,Ph),7.37(2H,t,J=3.0 Hz,Py),7.36(4H,d,J=6.0 Hz,Ph),7.12(8H,d,J=3.0 Hz,Ph)。IR(cm-1):3 060(νC—H),1 625,1 578,1 476,1 463,1 279(νC=C,νC=N,νC—N),1 243,1 279(νPh—O),833,777,739(δC—H)。
配体BL2的合成:称取0.41 g(1 mmol)2,2′-二(4-氨基苯氧基)苯基丙烷和0.54 g(5 mmol)2-吡啶甲醛,加入35 mL甲醇,加热回流4.5 h。反应结束后,用旋转蒸发仪将溶液蒸至5 mL,然后加入15 mL石油醚,待温度冷却到0℃,有黄色固体析出。过滤,真空干燥得到BL2固体0.42 g,产率为72%。元素分析(C39H32N4O2)计算值(%):C,79.6;H,5.5;N,9.5。实验值:C,79.6;H,5.4;N,9.6。1H NMR(CDCl3):δ8.80(2H,s,Py),8.64(2H,s,CH),8.19(2H,d,J=6.0 Hz,Py),7.81(2H,t,J=9.0 Hz,Py),7.54(4H,d,J=6.0 Hz,Ph),7.37(2H,t,J=3.0 Hz,Py),7.36(4H,d,J=6.0 Hz,Ph),7.12(8H,d,J=3.0 Hz,Ph),1.70(6H,s,CH3)。IR(cm-1):3 060(νC—H),1 625,1 578,1 476,1 463,1 279(νC=C,νC=N,νC—N),1 243,1 279(νPh—O),833,777,739(δC—H)。
配体BL3的合成:称取0.44 g(1 mmol)4,4′-二(4-氨基苯氧基)二苯砜和0.54 g(5 mmol)2-吡啶甲醛,加入35 mL甲醇,加热回流4.5 h,反应结束后,用旋转蒸发仪将溶液蒸至5 mL,有黄色固体析出。过滤,真空干燥得到0.51 g固体BL3,产率为83%。元素分析(C36H26N4O4S)计算值(%):C,70.8;H,4.3;N,9.2。实验值(%):C,70.7;H,4.3;N,9.3。1H NMR(CDCl3):
δ8.64(2H,d,J=3.0 Hz,Py),8.23(2H,s,CH),7.98(2H,d,J=6.0 Hz,Py),7.86(4H,d,J=6.0 Hz,Ph),7.83(2H,t,J=9.0 Hz,Py),7.41(4H,d,J=6.0 Hz,Ph),7.36(8H,d,J=3.0 Hz,Ph)。IR(cm-1):1 629,1 585,1 488,1 296(νC=C,νC=N,νC—N),1 243(νPh—O),833,777,742(δC—H)。
配合物Ru1的合成:称取0.52 g(1 mmol)Ru(bpy)2Cl2·2H2O和0.34 g(2 mmol)AgNO3,放入250 mL圆底烧瓶中,加入100 mL乙醇,磁力搅拌加热回流2 h,趁热过滤,除去AgCl沉淀,得到棕红色滤液A。称取0.27 g(0.5 mmol)配体BL1于250 mL三颈烧瓶中,将滤液A加入烧瓶中,反应液在N2保护下回流搅拌12h。停止反应,旋转蒸发至有固体出现,以CH3CN/饱和KNO3水溶液(1∶1,V/V)的混合溶剂作为洗脱剂,在硅胶柱上分离,得到的黄色溶液通过旋转蒸发除去大部分溶剂,然后加入过量的NaClO4的饱和乙醇溶液,沉淀出黄色固体,过滤,用热的甲醇和水(10 mL)各洗3次,真空干燥,得到黄色固体0.63 g,产率72%。,元素分析(C76H58N12O18Cl4)计算值(%):C,51.5;H,3.3;N,9.5。实验值(%):C,51.6;H,3.4;N,9.7。1H NMR(DMSO-d6):δ8.89(2H,t,J=3.0 Hz,Py),8.69(2H,d,J=6.0 Hz,Py),8.64~8.67(2H,m,Py),8.51(2H,t,J=6.0 Hz,bipy),8.21~8.24(6H,m,bipy),8.01(4H,t,J=9.0 Hz,Py),7.86(2H,d,J=3.0 Hz,Py),7.79(2H,s,CH),7.72(12H,m,bipy),7.50(4H,d,J=6.0 Hz,Ph),7.38(4H,d,J=9.0 Hz,Ph),6.80~7.00(4H,m,bipy),6.69(4H,d,J=3.0 Hz,bipy),6.64(4H,d,J=3.0 Hz,bipy)。IR(cm-1):1 603,1 489(νC=C,νC=N),1 240(νPh—O,νC—N),768,625(δC—H),1 106(νCl—O)。
配 合 物Ru2的 合 成:0.29 g(0.5 mmol)BL2和0.52 g(1 mmol)Ru(bpy)2Cl2·2H2O在乙醇溶液里回流12 h,硅胶柱分离,以乙腈/硝酸钾的饱和溶液(1∶1,V/V)为洗脱剂,得到黄色产品0.65g,产率74%。元素分析(C79H64N12O18Cl4)计算值:C,52.3;H,3.5;N,9.3。实验值(%):C,52.4;H,3.9;N,9.1。1H NMR(DMSOd6):δ8.89(2H,t,J=3.0 Hz,Py),8.66(2H,d,J=6.0 Hz,Py),8.62(2H,d,J=6.0 Hz,Py),8.49(2H,d,J=6.0 Hz,bipy),8.24~8.28(8H,m,bipy),7.96~8.00(4H,m,Ph),7.86(2H,s,CH),7.79(2H,d,J=9.0 Hz,Py),7.69(6H,m,bipy),7.64(4H,m,bipy),7.54(4H,d,J=6.0 Hz,Ph),7.35(4H,d,J=3.0 Hz,Ph),7.27(4H,d,J=9.0 Hz,Ph),6.76(6H,d,J=3.0 Hz,bipy),6.67~6.70(6H,m,bipy),1.65(6H,s,CH3)。IR(cm-1):1 625,1 578,1 476,1 463,1 279(νC=C,νC=N,νC—N),1 243,1 279(νPh—O),833,777,739(δC—H),1 118(νCl—O)。
配 合 物Ru3的 合 成:0.30 g(0.5 mmol)BL3和0.52 g(1 mmol)Ru(bpy)2Cl2·2H2O在乙醇溶液里回流12 h,硅胶柱分离,以乙腈/硝酸钾的饱和溶液(1∶1,V/V)为洗脱剂,用NaClO4的饱和乙醇溶液沉降,得到黄 色 产 品0.75 g,产 率82%。元 素 分 析(C76H58N12SO20Cl4)计算值(%):C,49.7;H,3.2;N,9.2。实验值(%):C,49.7;H,3.2;N,9.2。1H NMR(DMSOd6):δ8.87(2H,t,J=3.0 Hz,Py),8.67(2H,d,J=6.0 Hz,Py),8.62(2H,d,J=6.0 Hz,Py),8.50(4H,t,J=9.0 Hz,bipy),8.22~8.29(8H,m,bipy),8.01(4H,d,J=6.0 Hz,Ph),7.96~7.99(4H,m,bipy),7.86(2H,d,J=9.0 Hz,Py),7.79(2H,d,J=3.0 Hz,CH),7.72(4H,d,J=9.0 Hz,bipy),7.68(4H,d,J=6.0 Hz,Ph),7.64(4H,d,J=6.0 Hz,Ph),7.53(4H,d,J=3.0 Hz,Ph),7.35(2H,t,J=9.0 Hz,bipy),6.90(2H,d,J=6.0 Hz,bipy),6.84(4H,d,J=9.0 Hz,bipy),6.69(4H,d,J=3.0 Hz,bipy)。IR(cm-1):1 603,1 583,1 487,1 465,1 294(νC=C,νC=N,νC—N),1 244(νPh—O),1 108(νCl—O),768,626,558(δC—H)。
1.3 生物活性检测
按照标准的MTT测试方法进行活性检测[20-21],分为4步:(1)种入细胞。在96孔板的第3~11列用排枪分别加入悬有细胞的200 μL培养基,第2列作为溶剂空白,96孔板周围加培养基防止挥发,使每孔细胞数大约为1×104个,将96孔板放入37℃、CO2体积分数5%的培养箱中过夜,观察细胞生长情况,待80%细胞贴壁后,进行下一步。(2)加药。在24孔板的10个孔中各加入3 mL培养基,将所加药物稀释到不同浓度梯度,用排枪将孔中原有的培养液吸掉,每一列加入相同浓度的药物,加完后,放入培养箱培养48 h。(3)加MTT。在药物与细胞相互作用结束前4 h,吸去培养液,用磷酸缓冲盐溶液(PBS)洗涤2次,然后每孔加入180 μL PBS,再加入20 μL 5 mg·mL-1的MTT,放入培养箱继续培养4 h。(4)加DMSO。4 h后拿出96孔板,吸掉上清液,每孔加入150 μL DMSO,放入摇床内摇10 min,将紫色结晶物充分溶解,放入酶标仪卡槽内,在490 nm波长处扫描,得到光密度(OD),OD可以间接地反映活细胞的数量(式1),以此来求出半数抑制浓度IC50。
R=ODexp/ODcontrol×100% (1)
其中,R表示细胞存活率(即细胞活力),ODexp表示实验组OD,ODcontrol表示对照组OD。
2 结果与讨论
2.1 配体的合成
通过2-吡啶甲醛和对应的二元胺以2∶1的物质的量之比在甲醇中加热回流,可以得到黄色固体,用热的甲醇洗涤3次,得到黄色产品。元素分析得到的测试值与计算值基本符合,3个配体的红外光谱图中在1 625 cm-1处均有吸收带,说明配体中有C=N键 存 在,以CDCl3为 溶 剂,对BL1~BL3进 行了1H NMR表征。BL1的1H NMR谱图中有8组峰(图S1,Supporting information),苯 环 上Ha和Hb的δ=7.12,是双重峰;另一个苯环上Hc和Hd的δ为7.36和7.54,是2组双重峰;—CH=N氢的δ=8.64,是一个单峰;吡啶 环 上氢的δ分别为7.37、7.81、8.19和8.80,Hg和Hh是三重峰,Hf和Hi是双重峰。由于py-CH=N是一个吸电子基团,Hc和Hd离py-CH=N近,所以Hc和Hd的化学位移与Ha和Hb相比向低场移动。BL2和BL3的1H NMR谱图与BL1相似。
2.2 配合物的合成
配合物Ru1~Ru3分别通过Ru(bpy)2Cl2·2H2O和相应的配体BL1、BL2和BL3以2∶1的物质的量之比反应,用NaClO4饱和溶液沉降出产物,元素分析结果表明,所生成的配合物是双核配合物。配合物Ru1~Ru3的1H NMR在DMSO-d6中测 定,配 合 物Ru3的1H NMR谱图如图3所示。金属配合物的图谱相比配体较复杂,一是因为芳香环上氢的电子云密度相近,以至于它们的化学位移接近,芳香环区域光谱比较复杂,很难指认是哪个氢的化学位移;另一方面,因为金属离子与配体反应时生成一对对映体,所以配合物的核磁图谱比较复杂。但是,3个配合物在δ=9.0处都有一个明显的单峰,这个峰对应CH=N上氢的化学位移。
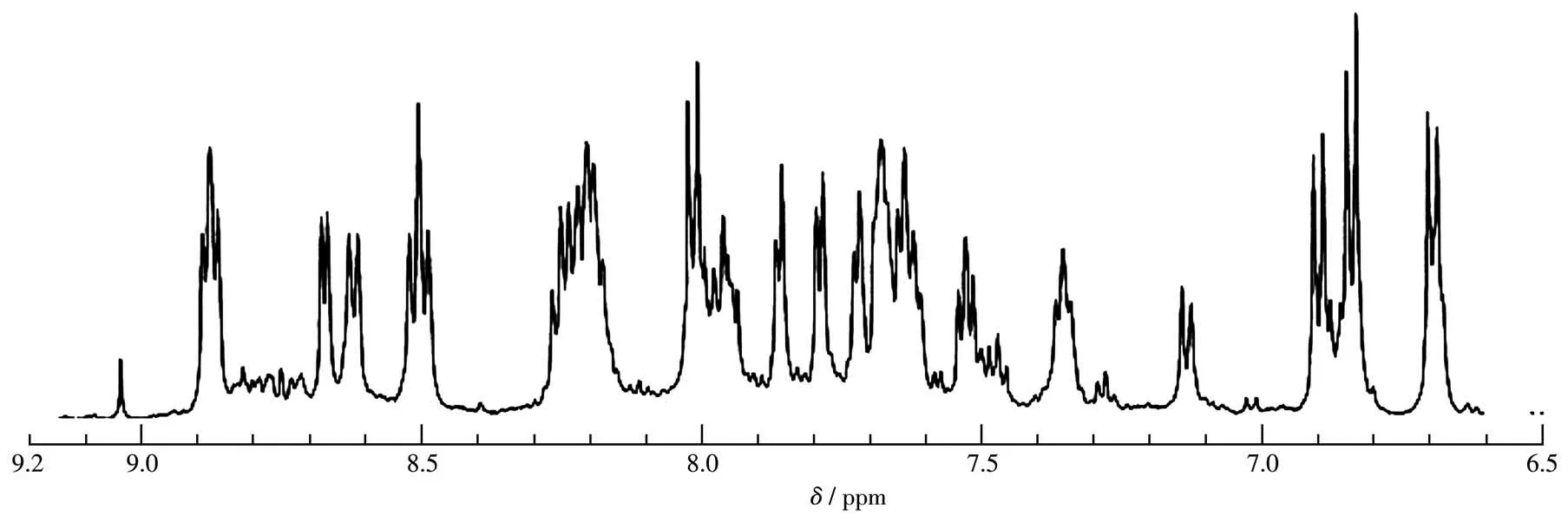
图3 配合物Ru3在DMSO-d6中的1H NMR谱图Fig.3 1H NMR spectrum of complex Ru3 in DMSO-d6
为了进一步确认配合物的结构,以CH3CNCH3OH为溶剂,测定了配合物的电喷雾质谱。电喷雾质谱对于证明分子量比较大的过渡金属配合物的分子量是非常可靠的。3个配合物的MS谱图中都只有一个强的离子峰,它们的m/z值分别为343.6([Ru(BL2)(bpy)2]24+)、354.2([Ru(BL1)(bpy)2]24+)和359.3([Ru(BL3)(bpy)2]24+),与计算值相符(图S2)。这一结果表明双核钌配合物在溶液中是稳定的。
2.3 配合物的电化学性质
采用微分脉冲伏安法(differential pulse voltammetry,DPV)来测定配合物的氧化还原电位,将配合物溶解在乙腈里,进行DPV测试,数据列于表1中。3个配合物在0~1.5 V范围内均有2个氧化还原峰,而在外界条件相同的情况下测定配体的氧化还原电位,发现配体在0~1.5 V内没有明显的峰,所以这些峰为金属中心的氧化-还原峰。从图4可以看出,Ru3的2个氧化还原峰分别在0.90和1.28 V,分别对应于[Ru(BL3)(bpy)2]25+/[Ru(BL3)(bpy)2]24+和[Ru(BL3)(bpy)2]26+/[Ru(BL3)(bpy)2]25+电对,2个半波电位的差值为380 mV。
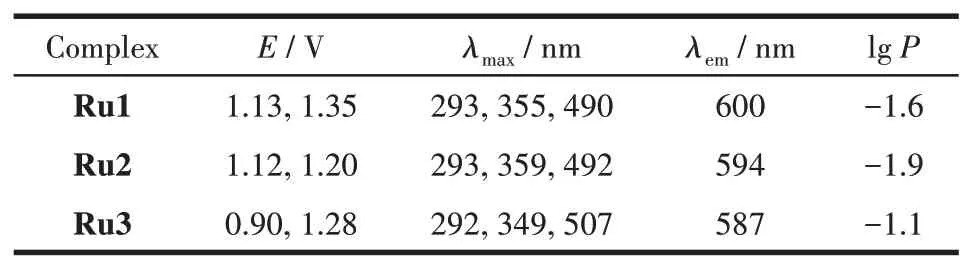
表1 Ru1~Ru3的光谱学、电化学数据及脂水分配系数(lg P)Table 1 Spectroscopic,electrochemistry data and fat-water partition coefficient(lg P)of Ru1-Ru3
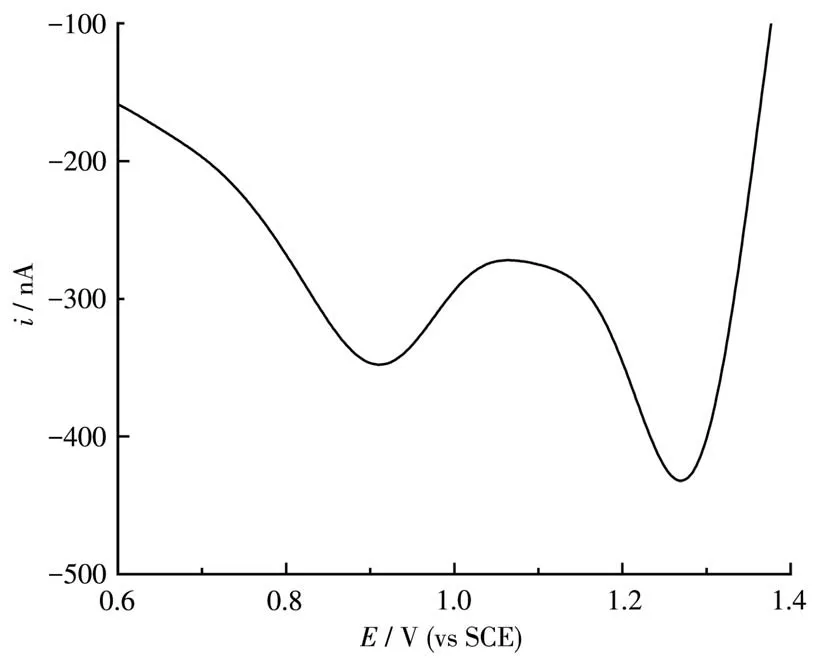
图4 配合物Ru3在CH3CN中的DPV图Fig.4 DPV diagram of complex Ru3 in CH3CN
2.4 配合物的光谱性质
将3个配合物分别配制成10 μmol·L-1的乙腈溶液,测定其UV-Vis光谱,所得谱图如图5a所示。表1列出了3个配合物Ru1、Ru2和Ru3的最大吸收波长。3个配合物的UV-Vis光谱相似,均有3个吸收峰,各个配合物对应的最大吸收峰波长值非常接近,分别在290、360和490 nm附近。每个吸收峰都可以找到相应归属:290 nm对应为一个又窄又强的吸收带,归属为未带任何取代基的联吡啶的π-π*电子跃迁产生的吸收;360 nm对应于电子从金属跃迁到配体的单重态吸收,即为1MLCT吸收;490 nm对应的较宽谱带归属为电子从金属跃迁到配体的三重态吸收,即为3MLCT吸收。
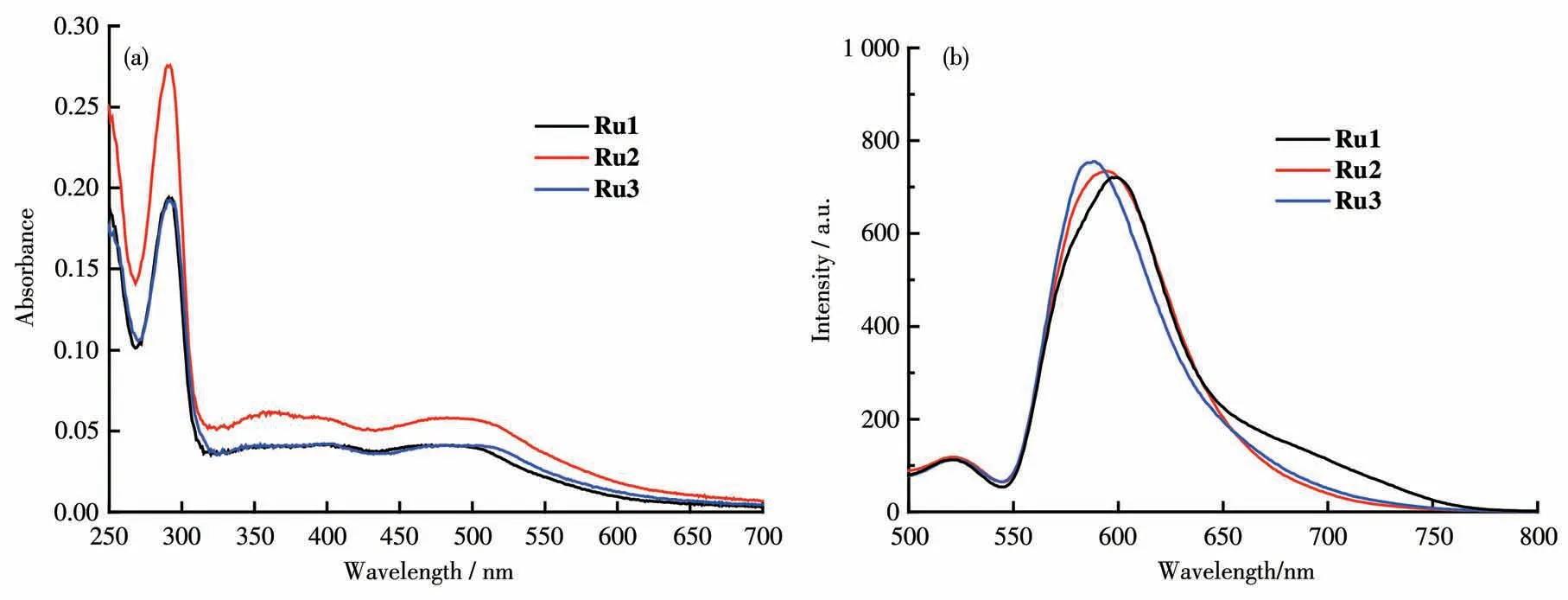
图5 配合物Ru1~Ru3在CH3CN中的(a)UV-Vis和(b)荧光光谱图Fig.5(a)UV-Vis and(b)fluorescence spectra of complexes Ru1-Ru3 in CH3CN
将配合物溶解在乙腈中,配成浓度为10 μmol·L-1的溶液,对配合物进行荧光光谱测试,以450 nm作为激发波长,扫描发射光谱,结果如图5b所示。3个配合物的发射波长均在600 nm左右,配体没有荧光,说明该系列配合物的荧光是金属钌发射出来的。
2.5 生物活性检测
以人宫颈癌细胞Hela、胃癌细胞BGC823、胃癌细胞SGC-7901和人正常胚肺成纤维细胞MRC-5为对象,通过MTT法检测了3个配合物对肿瘤细胞的细胞毒性。配合物对各细胞作用48 h后能够看到较明显的肿瘤细胞死亡变化。图6显示了配合物Ru1~Ru3对Hela、BGC823、SGC-7901和MRC-5的细胞毒性结果。
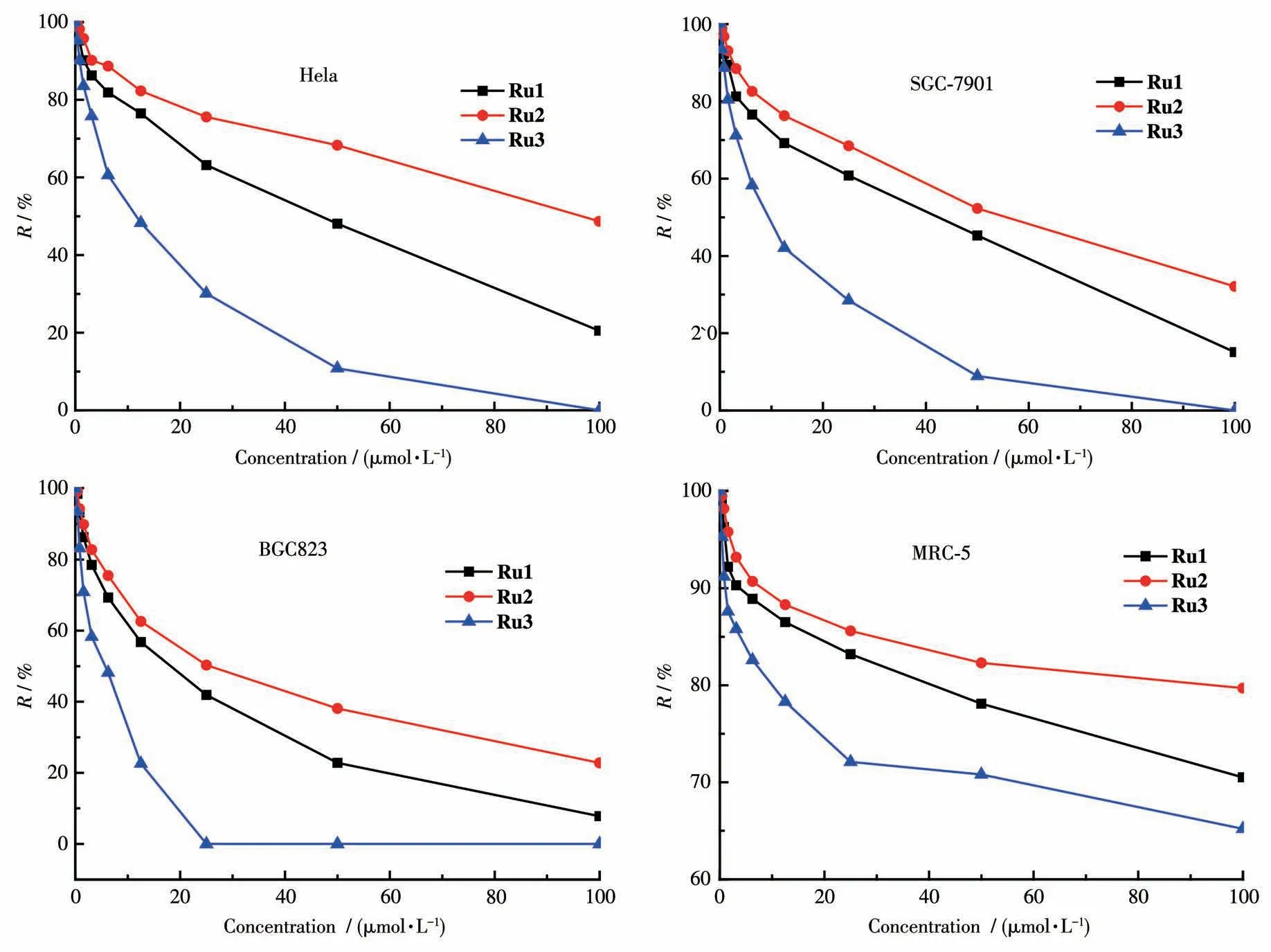
图6 配合物Ru1~Ru3对Hela、BGC823、SGC-7901和MRC-5的细胞毒性Fig.6 Cytotoxicity of complexes Ru1-Ru3 on Hela,BGC823,SGC-7901,and MRC-5
从表2可以看出,3个配合物对3种肿瘤细胞的毒性大小排序均为Ru3>Ru1>Ru2,可能的原因是配合物Ru3含有亚砜结构,配合物疏水性强,亲脂性大,溶解度低,使得配合物容易累积到线粒体内[22]。还可以看到,该系列配合物对BGC823的毒性均为最强。而3个配合物对MRC-5的IC50都大于100 μmol·L-1,说明 配合 物对 正常 细胞 没有 细胞毒性。
2.6 配合物的脂水分配系数
抗癌药物的活性通常与它们的亲脂性有关,由此产生的疏水性可能有助于增加细胞对药物的摄取,从而增强其抗癌活性[23-27]。我们测试了Ru1~Ru3的标准辛醇-水分配系数(lgP),结果见表2。lgP的大小顺序与上面得到的配合物的细胞毒性强弱呈一致关系,也间接地证明了我们的推测有一定的道理。这一结果初步表明钌配合物的抗癌活性与lgP有直接关系,其抗癌活性随配合物亲脂性的增加而增强。
2.7 活性氧检测
通过MTT实验得出的结论是配合物对肿瘤细胞有毒性而对正常细胞没有毒性,为了证实这一点我们做了活性氧(ROS)检测实验。ROS是生物体代谢过程中产生的含氧自由基或者容易形成自由基的过氧化物的总称。肿瘤细胞内ROS水平要高于正常细胞,这使得抗氧化酶对ROS的耐受性达到了极限,如果ROS再升高则会导致肿瘤细胞的凋亡[28],即使ROS水平下降也对肿瘤细胞的增值造成负面影响。
将 配 合 物Ru1~Ru3分 别 作 用 于BGC823和MRC-5,每个配合物设置2个浓度(6.25和12.5 μmol·L-1)。培养24 h后用DCFH-DA作为荧光探针检测细胞内ROS的变化(图7)。结果发现配合物Ru1~Ru3能增加肿瘤细胞内ROS的水平,并且配合物浓度越高,增强作用越明显。配合物的作用强度为Ru3>Ru1>Ru2,与MTT实验结果一致。但是,配合物Ru1~Ru3对正常细胞内ROS的水平并没有明显的影响。
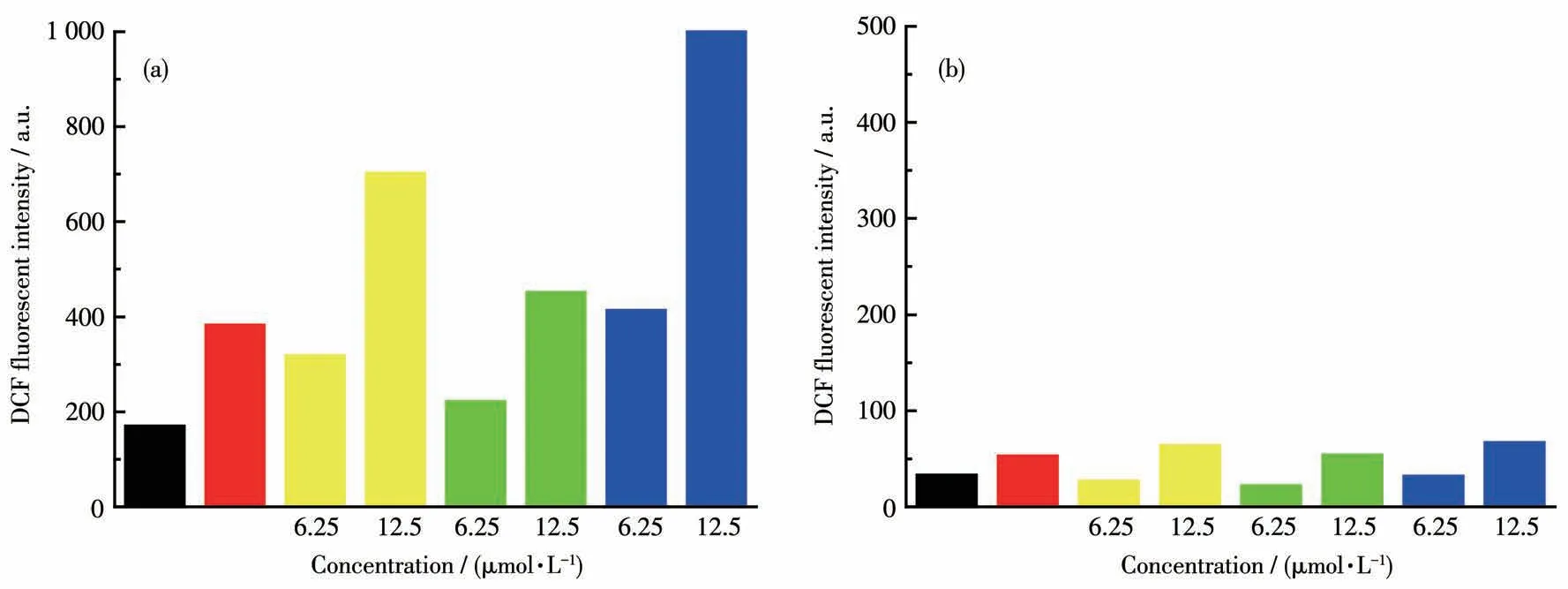
图7 配合物Ru1~Ru3对BGC823(a)和MRC-5(b)内ROS水平的影响Fig.7 Effect of complexes Ru1-Ru3 on ROS level in(a)BGC823 and(b)MRC-5
3 结论
以4,4′-二(4-氨基苯氧基)联苯、2,2′-二(4-氨基苯氧基)苯基丙烷、4,4′-二(4-氨基苯氧基)二苯砜和2-吡啶甲醛为原料,合成了3个双齿配体,并在此基础上合成了3个双核钌配合物Ru1~Ru3。通过元素分析、核磁共振氢谱、红外光谱、电喷雾质谱对配体及配合物进行了结构表征,证实了配合物结构的正确性。此外,通过MTT法测试了Ru1~Ru3对Hela、BGC823、SGC-7901和MRC-5四种细胞的毒性,结果显示Ru3的抗肿瘤活性强于Ru1和Ru2,而在所测的3种肿瘤细胞中,Ru1~Ru3均表现出对BGC823有很好的选择性(Ru3毒性最强),说明Ru3可能成为治疗癌症的潜在药物。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
农药学学报(2022年3期)2022-06-14
云南大学学报(自然科学版)(2022年3期)2022-05-25
节能与环保(2022年3期)2022-04-26
科学导报(2022年21期)2022-04-10
安徽农学通报(2022年6期)2022-04-07
北京大学学报(自然科学版)(2019年6期)2019-11-27
成长·读写月刊(2017年3期)2017-04-08
科技创新导报(2016年30期)2017-03-15
