
阿尔茨海默病脑TAU蛋白磷酸化位点综述
2022-10-19 12:28硕综述曹云鹏审校
中风与神经疾病杂志 2022年9期
刘 硕综述, 曹云鹏审校
阿尔茨海默氏病(AD)是痴呆中最典型的疾病,占所有痴呆病例的50%至60%[1]。AD的典型病理标志是神经元内神经原纤维缠结(NFT)和细胞外的淀粉样斑块(SP)沉积[2]。1988年,克劳德·维希克从AD患者大脑的斑块中分离出TAU,首次证明TAU可能是痴呆症的病因[3]。
TAU是神经元主要的微管相关蛋白,位于与微管相关的神经元轴突,可促进微管的组装以及稳定微管的结构[4]。TAU由17号染色体单基因编码,由于其前体基因的外显子2、3、10的选择性剪接,在成人大脑中表达为六种同工型,分别为2N4R(TAU441)、1N4R(TAU412)、0N4R(TAU383)、2N3R(TAU410)、1N3R(TAU381)和0N3R(TAU352),人类胎儿的大脑仅表达0N3R亚型[5]。根据序列和功能特性,可将TAU分为四个区域:NTR(N末端区域)、PRR(富含脯氨酸区域)、MBR(微管结合区域)以及CTR(C末端区域)[6]。
TAU是一种磷蛋白,正常低水平的磷酸化才能使TAU发挥最佳功能,而高水平异常磷酸化的TAU则失去其生物学活性[7]。Herrmann等用ELISA测定快速尸解的AD与同年龄正常人的脑组织匀浆,表明 AD组的额叶皮层、顶叶皮层和海马的TAU磷酸化水平明显增加,其中海马最高,但在小脑中未观察到差异[8]。在正常的人脑中,每摩尔TAU含有2~3 mol磷酸盐,且负面调节TAU与微管的结合,而在AD脑中,TAU被3~4倍过度磷酸化,每摩尔TAU含有8 mol磷酸盐,磷酸化水平达到饱和[9],并聚成神经原纤维缠结(NFT)[10]。NFT计数与TAU磷酸化水平正相关[8]。TAU的C末端区域的高水平磷酸化可能是其自聚集为NFT的原因[11]。NFT结构中有大量异常的双股螺旋纤维(PHF),成束密集排列,富含超磷酸的TAU[12]。认知障碍在经典缠结形成之前发生,TAU过度磷酸化可能是AD认知障碍的风险机制[13]。
1 AD患者TAU磷酸化位点
在由441个氨基酸组成的最长的人类TAU亚型中,总共有85个可磷酸化位点,其中45个丝氨酸残基,35个苏氨酸残基和5个酪氨酸残基,分别占TAU可磷酸化残基的53%、41%和6%[14],5个酪氨酸残基为Tyr18,Tyr29,Tyr197,Tyr310,Tyr394[15]。单个TAU分子不可能在所有这些位点都被磷酸化,PHF中 TAU至少在37个丝氨酸/苏氨酸残基位点及2个酪氨酸残基位点磷酸化,包括:Thr-39、Ser-46、Thr-69、Thr-123、Ser-137、Thr-153、Thr-175、Thr-181、Ser-198、Ser-199、Ser-202、Thr-205、Ser-208、Ser-210、Thr-212、Ser-214、Thr-217、Thr-231、Ser-235、Ser-237、Ser-238、Ser241、Ser-262、Ser-285、Ser-305、Ser-324、Ser-352、Ser-356、Ser-396、Ser-400、Thr-403、Ser-404、Ser-409、Ser-411、Ser-412、Ser-413、Ser-416、Ser-422、Thr-427、Ser-433、Ser-435[7]以及Ser-68、Thr-71、Ser-113、Ser-184、Ser-185、Ser-191、Tyr-197、Ser-258、Tyr-394、Thr-427、Ser-433、Ser-435[16]。其中最主要的十个超磷酸化位点是Ser198,Ser199,Ser202,Thr231,Ser235,Ser396,Ser404,Ser409,Ser413和Ser422[17],Ser404磷酸化程度最高[18]。在AD大脑中,TAU过度磷酸化会诱导恶性循环,促进TAU病理在脑中的传播以及在人脑脊液(CSF)中的积累[19]。Russell及Mitra等通过质谱法对CSF中的磷酸化TAU分析表明,与正常人脑对照组相比,AD患者的CSF中一些位点磷酸化明显增加(见表1)。但AD患者CSF Ser202磷酸化程度却较正常人低[18]。
在AD早期磷酸化程度就升高的位点:Thr181,Ser199,Thr205和Ser404[20]Thr217,Ser262,Thr231、Ser396、Thr212[21,22],其中TAU的Ser199残基在AD发展的早期和晚期均异常过度磷酸化[23],Thr231的磷酸化程度随AD的发展而降低[24]。Ser202、Thr205、Ser409和Ser422磷酸化程度升高发生在NFT后期[25]。
异常高磷酸化的TAU可以隔离正常TAU与微管,进一步破坏微管动力学,但需要多个位点同时磷酸化才能破坏TAU与微管之间的相互作用[26]。Thr212,Ser214,Thr231,Ser235和Ser262,是抑制TAU结合微管的主要位点,Ser262、Thr231、Ser 235处磷酸化大大降低了Tau与微管结合的能力分别降低了33%、26%和9%,导致微管不稳定[27];在AD神经退行性变早期,Thr212,Thr231和Ser262这三个磷酸化升高位点的组合将TAU转化为毒性分子,导致微管网络的破坏和细胞死亡[22];磷酸化的Thr231是对去磷酸化具有异常抵抗力的残基[26],磷酸化水平升高可预测轻度认知障碍患者向AD转化[28];磷酸化Thr181是CSF中公认的AD核心生物标志物[29];TAU在Ser396和Ser404处的磷酸化增加通过从微管中分离正常TAU分子而损害微管组装,在体外产生更多的原纤维形式TAU,聚集增加[1];Ser202、Thr205磷酸化增加使得TAU微管成核活性降低,微管解聚和不稳定,同时与后来转化为原丝和细丝有关[30];Thr205的磷酸化可特异性抑制淀粉样蛋白β(Aβ)的毒性,是具有保护作用而不是毒性促进作用的唯一位点[31];Ser199的磷酸化是AD中,脆弱的神经元向神经纤维变性失衡的关键步骤[23]。Yu等研究表明Thr181,Thr205,Thr217和Ser404磷酸化在大鼠胚胎期和出生后早期在神经突生长中起关键作用,Ser202,Thr212,Ser356和Ser404的磷酸可能与神经突生长无关[11]。所有这些位点的磷酸化也不是完全独立的,如必须先磷酸化Ser235才能使Thr231磷酸化[26];Ser214处的磷酸化显着抑制了随后在Thr212和Thr217处的磷酸化,;Ser409处的磷酸化抑制了随后在Ser404处的磷酸化[32]。
2 正常成人TAU磷酸化位点
对人脑组织活检的研究表明,正常人脑TAU在大多数与PHF-TAU相同的位点上以低计量水平被磷酸,病理上这些位点则磷酸化过度[7,33]。由于检测方法的不同,所检测到的正常人脑TAU的磷酸化位点也不同。这些位点包括活检法检测到位点:Thr-181、Ser-202、Ser-214、Thr-231、Ser-262、Ser-396、Ser-404、Ser-409[26,34~36];质谱法检测到的位点:Thr-50、Thr-52、Thr-56、Ser61/Thr63/er64(至少两个点)、Ser-68、Thr-69、Thr-71、Thr-76、Thr-101、Thr-102、Thr-111、Ser-113、Thr-123、Thr-175、Thr-181、Ser-184、Ser-185、Ser-198、Ser-199、Ser-202、Thr-212、Ser-214、Thr-217、Thr-231(不确定)、Ser-396、Ser-404、Ser-409、Ser-411、Ser-412、Ser-413(Ser411/Ser412/Ser413至少2个点)、Ser-416[37];IIPSC法检测到的位点:Thr-181、Ser-198、Ser-199、Ser202/Thr205、Thr-231、Ser-262、Ser-396、Ser400/、Thr403/、Ser404Ser-416[1,38]。一定水平的磷酸化是正常的,有助于神经元维持其微管网络和细胞活性。Barthélemy R等用质谱法检测正常人CSF中TAU磷酸化位点,如表1所示,但是与AD患者相比,磷酸化水平较低。
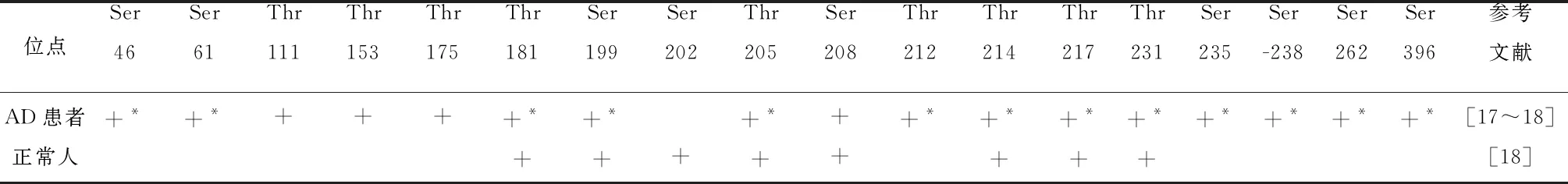
表1 CSF中磷酸化位点
在人脑的天然TAU中,Ser422磷酸化程度非常弱或完全不被磷酸化[35]。也有其他研究方法显示正常成年人大脑中TAU的Ser195、Ser198、Ser199、Thr212位点不会被磷酸化,Ser199位点在儿童及青年期被磷酸化,但随着年龄的增长磷酸化水平降低[23]。
Thr172,Ser193,Thr222,Ser387和Ser395是大鼠脑TAU中磷酸化的正常位点[39],正常大鼠TAU在Thr212,Thr217,Thr231,Ser396,Ser404,Ser422的磷酸化非常低,而在AD大鼠中的磷酸化增加到10到100倍[11]。
3 胎儿脑TAU磷酸化位点
与正常成人脑TAU相比,胎儿脑TAU磷酸化程度更高,但没有明显的毒性[28,34]。成年大脑整体TAU磷酸化水平的降低可能是由于磷酸酶水平的升高,以及激酶水平下调[11,23,39]。
TAU的表达和磷酸化水平均受发育调节。高磷酸化的TAU是发育过程中轴突发生所必需的,对微管可塑性具有必要性[36]。发育中大脑的TAU可能比成熟大脑的TAU具有更小的微管结合活性[11]。与成人脑TAU相比,胎儿脑TAU在与AD脑几个相同的磷酸化位点上被高度磷酸化,如表2所示,但Ser 409位点高度磷酸化似乎是AD特有的[36]。
Hefti等在大鼠中也有相似发现,但与人脑不全相同,如表2所示。AD大鼠大脑Ser214位点的磷酸化水平低于或与胎儿大鼠大脑相似,但人胎儿大脑中Ser214位点的磷酸化程度比AD患者要强得多[38]。Ser262和Ser356,位于微管结合域内,可以认为Ser262和Ser356位点的磷酸化对发育大脑中TAU的生物学活性至关重要[11]。
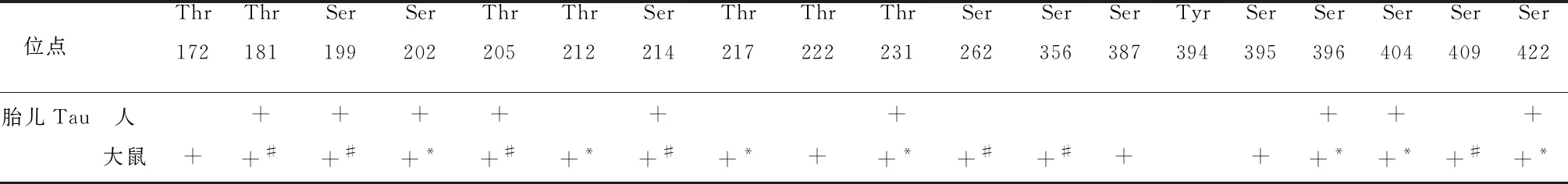
表2 人与大鼠胎儿Tau磷酸化位点
尽管正常发育中的大脑和AD大脑的TAU都在多个位点被高度磷酸化,且在前者中发现了无毒的TAU聚集体[36],但只有后者抑制了微管的组装,并聚合成NFT[11],正常胎儿大脑TAU的高度磷酸化不足以诱导神经原纤维病理。
4 FAD与SAD的TAU磷酸化位点
大多数AD病例是散发性的,病因不明,称为散发性阿尔茨海默氏病(SAD),只有5%的AD患者表现出该病的家族性单基因形式,称为家族性阿尔茨海默氏病(FAD)[40]。在FAD病例中,约有一半与早老素1(PS-1)基因突变有关,而早老素2(PS-2)和淀粉样蛋白前体蛋白(APP)基因突变代表较少[41]。FAD早期发病,迅速发展到终末期期[42]。有证据显示PS-1突变加速NFT形成,这表明PS-1突变也可能影响TAU磷酸化[7],但PS-1突变的多态性不影响与AD发生和发展有关的最终神经病理学,FAD和SAD之间的总病理负荷没有差异[42]。
在FAD和SAD患者诱导多能干细胞(iPSC)系衍生的神经元中比较TAU超磷酸化的研究显示,Ser262、Ser396、Ser202 / Thr205、Thr181、Ser198、Ser199、Thr231、Ser400 / Thr403 / Ser404、Ser416位点磷酸化水平在FAD和SAD衍生的神经元之间未观察到主要差异[1,38]。
与年龄匹配的野生型同窝仔(WTs)相比,表达PS-1(L235P)的老年(21月半龄)转基因小鼠海马中,TAU在Ser396,Ser404,Thr231和Ser198 / 199/202表位的磷酸化显着增加[43]。
5 尸检与活检
从活检到尸检,AD患者SP和NFT的密度以及小胶质细胞活化显着增加[42]。尸检的AD患者大脑TAU磷酸化水平降低,很大程度上归因于磷酸酶的能量非依赖性活性[43],长期的尸检间隔时间(PMI)可能会低估TAU磷酸化水平[44]。活检来源患者大脑TAU不会因组织中的内源性磷酸酶而经历极快的去磷酸化作用[25]。但是相较于正常人,AD患者死后验尸延迟期间的磷酸化位点迅速脱磷酸化程度低,这可能是由于蛋白质磷酸酶活性不足以及TAU的聚集所致[7]。
6 总 结
AD是一种复杂的多因素疾病。AD典型的病理标志是NFT和细胞外的SP。多达30%的正常老年人大脑中的Aβ斑块负荷与AD的典型病例一样多,但是他们缺乏Aβ斑块周围的TAU病理性营养不良性神经突[45]。免疫疗法是在AD治疗领域最有希望的方法之一。但是,针对Aβ的免疫疗法临床试却验接连失败,主要原因之一是大脑中的Aβ负荷与痴呆程度无关。靶向TAU的免疫疗法是针对Aβ免疫疗法失败之后的新方向。AD病理毒性,主要是由于TAU过度磷酸化所致[46],所以对TAU磷酸化位点的研究,将会为更有效的治疗提供靶点。同时,AD患者CSF与大脑中的TAU磷酸化位点不全相同,CSF中TAU磷酸化位点研究将为AD诊断的生物标志物提供有意义的依据。
不同的实验研究对象及方法会出现不同的实验结果。鼠和人脑TAU磷酸化位点不全相同;由于磷酸激酶与磷酸酶的变化,相对于活检,尸检TAU磷酸化位点减少;Western、Elisa、免疫组化、Dot-Blot等实验方法的不同,也会使得实验结果有偏差。现在,质谱法、ipsc等方法为研究TAU磷酸化位点提供新的思路,TAU磷酸化位点的研究前景也将会越来越明朗。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
学苑创造·A版(2022年3期)2022-03-29
电脑报(2020年40期)2020-11-06
中国医药导报(2019年7期)2019-05-13
电脑知识与技术(2018年19期)2018-11-01
科教导刊·电子版(2018年9期)2018-06-07
分析化学(2018年2期)2018-03-02
分析化学(2017年12期)2017-12-25
初中生世界·七年级(2017年5期)2017-06-10
