
基于微卫星复合扩增的马来穿山甲个体识别研究
2022-11-07 03:19李嘉昊逯金瑶
野生动物学报 2022年4期
李嘉昊 王 辰 雷 行 逯金瑶 李 立* 李 波,3*
(1.东北林业大学野生动物与自然保护地学院,哈尔滨,150040;2.湖南省生物多样性保护中心,长沙,410209;3.国家林业和草原局野生动植物检测中心,哈尔滨,150040)
据CITES的统计数据,2014—2018年缴获的穿山甲非法贸易个体数量超过14万只;另据UNODC的统计数据,2015年穿山甲肉类缉获量占比为15%,2016—2018年占比为1%~2%,但鳞片占比超过95%[3]。1998—2019年,马来穿山甲和菲律宾穿山甲的数量急剧下降,马来穿山甲的种群数量减少了约80%。穿山甲及其制品非法走私案件频发使穿山甲个体识别的法医学技术变得尤为重要。
目前,穿山甲个体识别由微卫星技术实现[4-5]。最早的微卫星位点是2007年基于马来穿山甲开发的34个双核苷酸重复位点[6],这为亚洲穿山甲个体识别提供了研究基础。高赛飞[4]从中选出6个多态性较高的位点,对50份穿山甲鳞片进行个体识别,认定其来自45只个体,但由于样本质量不高和未进行物种鉴定影响了个体识别的准确率。Sun等[7]从中筛选出10个高多态性位点,成功对54只中华穿山甲进行了基因分型,组合7个位点即可实现无关个体基因型匹配概率(probability of identity,P(ID))小于0.001,同胞个体基因型匹配概率(probability of identity for siblings,P(ID)sib)小于0.01[8]。Singh等[5]利用其中8个位点构建中华穿山甲和印度穿山甲个体识别体系(pangolin indexing system,PIS),使用细胞色素b(Cytb)基因确认待测样本的来源物种,排除了引物对物种适应度的影响。
目前亚洲穿山甲个体识别使用的是双核苷酸重复的微卫星位点,扩增过程中TaqDNA聚合酶的滑动容易导致双核苷酸重复位点出现影子峰而干扰基因分型,但使用较长重复单位的位点可减少影子峰[9]。同时,微卫星复合扩增能够提升鉴定效率,节约鉴定成本。根据国际法医遗传学会(International Society for Forensic Genetics,ISFG)对非人类个体识别的建议[10],需要优先使用四核苷酸重复、复合扩增方法和新引物标准化来重构马来穿山甲个体识别体系。
1 材料与方法
1.1 样本采集与DNA提取
收集中华穿山甲、马来穿山甲、印度穿山甲、树穿山甲、大穿山甲的肌肉、鳞片和皮肤样本总计156份,以及非穿山甲对照样本21份(表1),将样本置于冰箱内-20 ℃保存。样本预处理后利用蛋白酶K消化,后用基因组提取试剂盒(上海百赛生物工程技术股份有限公司)提取和纯化总DNA。使用紫外分光光度计(NanoPhotometer-N50,德国Implen)检测DNA浓度。
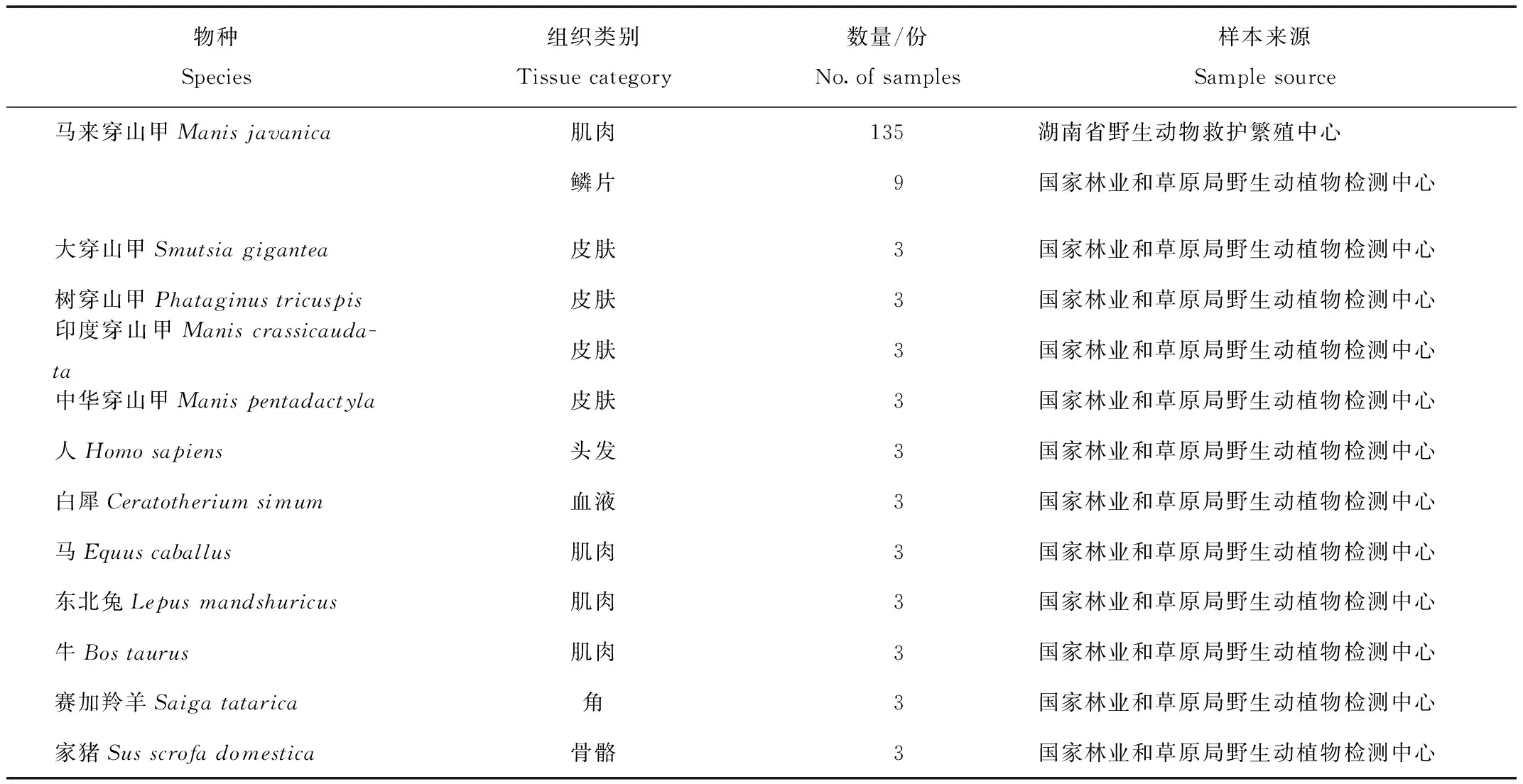
表1 试验样本信息
1.2 位点筛选和PCR扩增
使用的微卫星位点包括6个双核苷酸重复马来穿山甲位点[6];从长尾穿山甲和南非穿山甲中筛选的4个三、四核苷酸重复位点[11-12],其重复单元经测序已确认;以及利用SSRHunter软件从马来穿山甲基因组(GCF_014570535.1)[13]中筛选的四核苷酸重复位点(MJL04、MJL05),这2个位点经Primer premier 5.0软件设计成新引物后加入复合体系中(表2)。

PCR反应采用20.0 μL体系:2×EasyTaqSuperMix(TransGen,北京)10.0 μL,正、反向引物(10 pmol/μL)各0.2 μL,DNA模板2.0 μL,灭菌去离子水7.6 μL。正向引物分别用FAM、HEX和TAMRA荧光染料标记。扩增采用PE9700型DNA扩增仪(PE,美国)。扩增产物用1.5%琼脂糖凝胶电泳分离(90V,25 min),并以此检查所有引物的特异性。
1.3 复合扩增体系优化和评估
为优化复合扩增体系,使用梯度PCR测试最适退火温度(60~72 ℃设置15个梯度)。PCR扩增选择3个反应体积(40、30、20 μL)测试最适体系。PCR产物送生工生物工程(上海)股份有限公司进行毛细管电泳分型,根据毛细管电泳的信号峰强度调整和确定引物的最终浓度。
选用3份样本在3种不同的PCR仪器上用相同方案重复3次来测试复合体系的再现性。盲测试验时选用30份样本,当所选位点的P(ID)足够小(0.000 1~0.001 0)时,对比各自基因型,具有相同基因型的样本认定为同一个体。
利用DNA质量浓度梯度检测该体系的灵敏性和有效性。DNA质量浓度梯度分为20.0、10.0、5.0、2.0、1.0、0.5 ng/μL。在上述PCR条件下,将DNA稀释液添加到20.0 μL反应体系中复合扩增。利用7种非穿山甲物种(表1)评估该复合扩增体系的物种特异性。影子峰是双核苷酸重复位点基因分型分析中常见的现象,分析影子峰峰强度与目的产物峰强度的比例,可更准确地进行基因分型。
1.4 数据分析
使用GeneMarker 3.5进行基因分型,Cervus计算等位基因频率、等位基因数(number of allele,Na)、有效等位基因数(Ne)、观测杂合度(Ho)和期望杂合度(He)[14]。多态性信息含量(polymorphism information content,PIC)和哈迪-温伯格平衡(Hardy-Weinberg equilibrium,H-W)用Genepop 4.2计算[15]。基因流使用Popgene 3.2计算。用STRUCTURE分析马来穿山甲种群的遗传结构,建系方法与评判标准参照Pritchard等[16]与Evanno等[17]的研究。具体参数设置:种群个数(K)预设为1~10,每一个K都运行10次,初始不作数的迭代为10 000次,MCMC运行100 000次,利用在线软件STRUCTURE HARVESTER统计马来穿山甲种群的最佳K值。根据分型结果使用Population 1.2.32构建基于Nei氏遗传距离[18]的NJ系统发育树。
2 结果与分析
2.1 构建复合扩增体系
通过引物筛选、设计和PCR反应条件优化,获得12个微卫星位点的3个复合扩增体系(MP1、MP2和MP3;图1),每个位点的荧光产物峰强度大于1 000相对荧光单位(relative fluorescent units,RFU)。PCR扩增的最适程序:94 ℃预变性3 min;94 ℃变性45 s,56 ℃退火45 s,72 ℃延伸60 s,30个循环;94 ℃变性45 s,60 ℃退火45 s,72 ℃延伸60 s,25个循环;72 ℃延伸10 min。
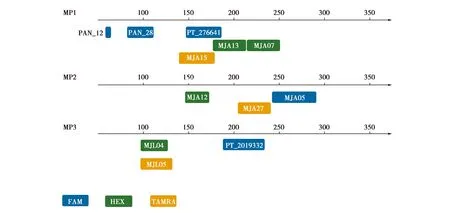
图1 复合扩增体系Fig.1 Multiplex amplification system
2.2 复合扩增体系的验证
12个微卫星位点在马来穿山甲种群(n=135)中共检出155个等位基因,所有等位基因组合成分型标准物。PAN_12和PAN_28位点的等位基因数量≤4个,而MJA05、MJA07、MJA13、MJA15和MJA27位点的等位基因数量均大于15个(附录1)。
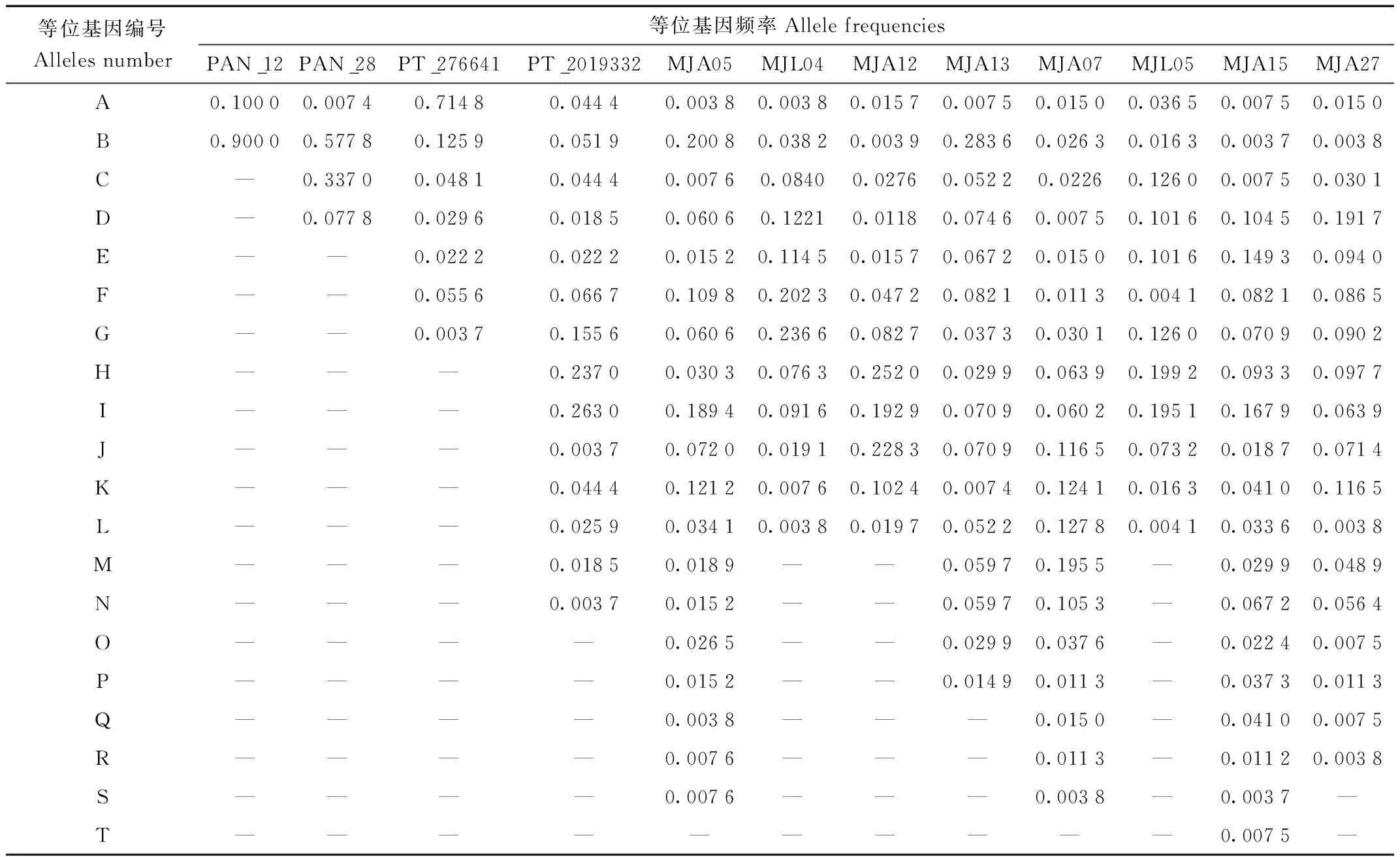
附录1 12个微卫星位点等位基因及基因频率
非父排除概率(probability of exclusion,PE)指非亲子关系能被遗传标记排除的概率。PE-1P指两个亲本的基因型未知时的排除概率;PE-2P指当一个亲本的基因型已知的排除概率;PE-PP指两个亲本的基因型均已知时的排除概率。12个位点的累计P(ID)为6.48×10-16,P(ID)sibs为9.65×10-6,累计PE为0.999 8~0.999 9(表3)。体系内的6个三、四核苷酸重复位点累积P(ID)为3.48×10-6,P(ID)sibs为9.7×10-3,便可满足法医非人类判别标准(P(ID)<0.001 0)[8]。

新构建的复合扩增体系在相同条件下进行重复性测试,所有马来穿山甲样本均能成功基因分型。在不同PCR仪器上扩增的每个样本在12个多态性位点上的基因型完全一致。盲测结果可以准确地将30份样本判定为源自20个不同个体,所有样本仅需1次分型,准确率为100%。
使用DNA浓度梯度进行灵敏度测试。当DNA质量浓度为0.5 ng/μL时,所有位点均能获得准确分型,最低峰强度为180 RFU,表明该复合扩增体系内所有位点能在微量的DNA检材中得到准确的分型结果(图2)。

图2 灵敏度测试Fig.2 Sensitivity test
对于穿山甲科其他物种,MJA12、MJL05和MJA15位点在树穿山甲样本中未检测到信号;所有位点在大穿山甲样本中检测到信号;MJA12和MJA15位点在中华穿山甲样本中未检测到信号。对于非穿山甲科物种,马(Equuscaballus)和白犀(Ceratotheriumsimum)有2个位点检测到信号;家猪(Susscrofadomestica)有3个位点检测到信号,牛(Bostaurus)、赛加羚羊(Saigatatarica)和东北兔(Lepusmandshuricus)都没有检测到信号(表4)。

表4 物种特异性分析
影子峰分析表明,三、四核苷酸重复位点相较于双核苷酸重复位点在基因分型时更加保守、可靠性更高(表5)。分析影子峰产生的概率和影子峰占目的峰强度的平均值,可有效筛除峰强度明显偏低的影子峰,从而最大限度消除影子峰对基因分型的影响。

表5 影子峰分析
2.3 种群遗传分析
12个位点共检测到155个等位基因(附录1),各位点有效等位基因(Ne)为1.219~10.602,平均值为6.462,其中PAN_12位点有效等位基因数量最少,MJA15位点有效等位基因数量最多。各位点期望杂合度(He)为0.180~0.912,其中PAN_12位点最小,MJA15位点最大。各位点观测杂合度(Ho)为0.140~0.822,其中PAN_12位点最小,MJA27位点最大(表6)。

表6 12个微卫星位点多态性
分析135只涉案马来穿山甲的种群遗传结构,结果如图3所示。随着假设种群数(K)增加,lnP(D)逐渐增加,而变化率逐渐减小,当K在2~3变化时幅度最大。当K=3时,lnP(D)的衍生值ΔK取最大值(25.6),结合lnP(D)的变化趋势和ΔK值,最终确定:K=3为最佳值,能反映待测种群的真实情况。将全部涉案个体划分为3个种群:MJ1、MJ2和MJ3。MJ3种群遗传结构较为单一,MJ1和MJ2种群则相对复杂,说明MJ1和MJ2种群间的基因交流相对较多,MJ3与其他种群间的基因交流较少。

图3 马来穿山甲结构聚类Fig.3 Structure cluster diagram of 135 Malay pangolins 注:红色表示MJ1种群;绿色表示MJ2种群;蓝色表示MJ3种群 Note:The red individuals are from the population of MJ1.The green individuals are from the population of MJ2. The blue individuals are from the population of MJ3
NJ系统发育树有类似结果(图4)。MJ2和MJ3种群形成了独立的亚支系,MJ1种群无独立的亚支系,但与MJ2种群个体混杂一起,表明MJ1与MJ2种群间遗传距离较近,MJ3与MJ1、MJ2种群间遗传距离相对较远。

图4 基于12个微卫星位点遗传距离构建NJ系统发育树Fig.4 NJ tree based on genetic distance of 12 microsatellite locus 注:红色表示MJ1种群;绿色表示MJ2种群;蓝色表示MJ3种群 Note:The red individuals are from the population of MJ1.The green individuals are from the population of MJ2.The blue individuals are from the population of MJ3
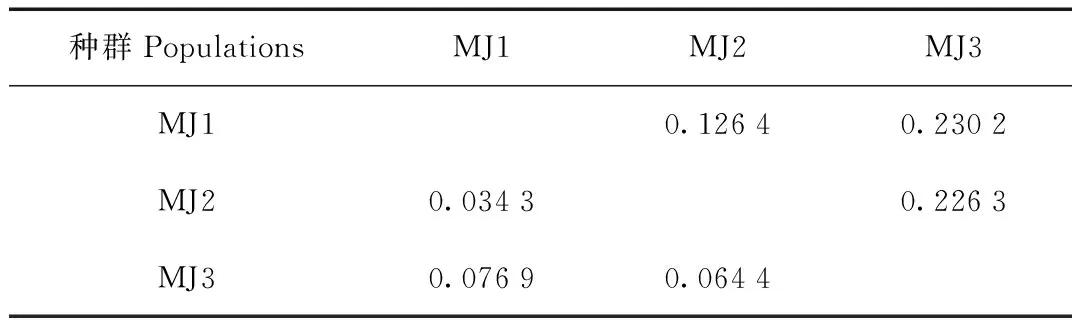
MJ1种群有2个位点显著偏离H-W平衡(P<0.01),10个位点符合H-W平衡(P>0.05)。MJ2种群有5个位点显著偏离H-W平衡(P<0.01),6个位点符合H-W平衡(P>0.05),1个位点偏离H-W平衡(0.01 附录2 哈迪-温伯格平衡检测 固定指数(fixation index,FST)可以体现种群间的遗传分化程度:FST<0.05表示弱分化,0.05≤FST≤0.25表示中度分化,FST>0.25表示高度分化。结果表明,MJ1和MJ2种群处于弱分化水平,MJ1和MJ2与MJ3种群分化程度较高,属于中度分化(表7)。MJ1和MJ2种群的遗传距离较近,二者与MJ3种群的遗传距离较远,这与NJ系统发育树的结果相同。 表7 MJ1、MJ2和MJ3种群间固定指数和种群间遗传距离 ISFG建议非人类个体识别时优先使用四核苷酸重复和复合扩增方法[10]。本研究筛选了6个双核苷酸重复位点和6个三、四核苷酸重复位点组成3个复合扩增体系(MP1、MP2和MP3)。尽管双核苷酸重复位点出现影子峰的概率较高,增加了基因分型的难度,但是分析影子峰峰强度与目的产物峰强度的比例,可以消除影子峰对分型的影响,提高基因分型的准确度[19]。同时,MP1体系相对于MP2和MP3灵敏度更高,包含三、四核苷酸重复位点更多,其基因分型的优势更大。若检材DNA质量不佳,推荐优先使用MP1进行个体识别鉴定。 本研究重构的马来穿山甲个体识别体系具有较高的鉴别力,其P(ID)值和P(ID)sibs值远大于已发表的个体识别系统[5,7]。该体系内6个三、四核苷酸重复位点组合便可满足非人类个体识别的法医学标准(P(ID)<0.001)[8]。物种特异性分析表明,中华穿山甲和大穿山甲样本中检测出10个位点以上的特异峰,说明该体系经大样本验证后可适用于中华穿山甲和大穿山甲的个体识别和亲缘关系鉴定。 鳞片是穿山甲非法贸易的主要部位,占比超过95%[3]。目前,在处理穿山甲鳞片非法贸易案件时,如何认定个体数量是一大难题,主要通过穿山甲鳞片干重来评估盗猎个体数量,但鉴定结果不准确。本研究重构的马来穿山甲个体识别体系可有效对鳞片样本进行个体识别,为解决上述难题提供了新方案。 本研究计算的马来穿山甲种群遗传多样性水平与前人的研究结果[4]相近,各位点He、Ho和PIC平均值的细微差异可能与选择的位点和样本数量差异有关(表8)。同时,马来穿山甲种群遗传多样性水平与印度穿山甲[5]类似,大于中华穿山甲[5-6]和非洲的4种穿山甲[11-12]。非洲4种穿山甲种群遗传多样性水平与其分布范围和种群数量呈正相关[12]。亚洲穿山甲中的中华穿山甲种群遗传多样性较低,应该与其濒危状况和种群数量密切关联。 表8 穿山甲科物种种群遗传多样性对比 Nash等[20]使用全基因组筛选的单核苷酸多态性位点(single nucleotide polymorphism,SNPs)分析了83只马来穿山甲的种群遗传结构,结果表明这些马来穿山甲可分为3个种群,分别来自婆罗洲、印度尼西亚爪哇岛和新加坡或苏门答腊岛。本研究的马来穿山甲种群遗传结构分析也得出了类似结果。但是,由于所用样本为非法贸易查获,无法追溯样本来源地,不能据此推测该批样本的原产地。依据非洲象牙的微卫星个体识别体系可用于象牙产地追踪[21],可在国际上推广利用本研究的马来穿山甲个体识别体系构建已知样本数据库,为非法贸易案件中穿山甲各类检材来源地的鉴定奠定基础。 总之,由12个微卫星位点重构的马来穿山甲个体识别体系具有可靠性强、准确率及灵敏度高和成本低等优点,可适用于多种类型的检材。同时,该体系可用于评估马来穿山甲种群遗传多样性和遗传结构。 致谢:本研究试验样本的采集得到了国家林业和草原局野生动植物检测中心的徐艳春教授、金煜教授、白素英副教授、马跃工程师、王震工程师和刘微工程师的大力支持与帮助,在此一并致谢。
3 讨论
3.1 位点筛选和个体识别体系重构
3.2 种群遗传分析
猜你喜欢
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
好孩子画报(2018年7期)2018-10-11
数学大王·中高年级(2017年12期)2017-12-21
四川动物(2017年6期)2017-12-12
小学生学习指导(低年级)(2017年11期)2017-10-23
四川动物(2017年4期)2017-07-31
幼儿100(2017年13期)2017-06-15
中学生(2017年13期)2017-06-15
小学生作文选刊·低年级版(2017年4期)2017-06-07
数学大王·中高年级(2016年10期)2016-09-10
