
三氟甲基磺酰氟绝缘介质理化特性的分子动力学模拟
2022-11-15 09:35张咪田亚锋高克利侯华王宝山
高等学校化学学报 2022年11期
张咪,田亚锋,高克利,侯华,王宝山
(1.武汉大学化学与分子科学学院,武汉 430072;2.中国电力科学研究院有限公司,北京 100192)
六氟化硫(SF6)气体具有优异的绝缘与灭弧性能,是高压、特高压电气设备中常用的绝缘介质.同时,SF6是一种极强的温室气体,其全球变暖潜势(GWP)约为CO2的23500倍.在双碳目标的驱动下,电气设备的环保化需求日益迫切,采用新型环保绝缘气体替代SF6势在必行[1].除了应用技术较为成熟的SF6/N2混合气体之外,一系列环境友好的新型替代气体[如三氟碘甲烷(CF3I)、全氟酮(C5F10O,C6F10O)、七氟异丁腈(C4F7N,简称C4)等]受到广泛关注[2,3].以上新气体的绝缘强度均优于SF6,但液化温度普遍较高,只能与缓冲气体(CO2,N2或空气)混合后用作电气设备的绝缘介质.受绝缘强度、液化温度、环保、毒性和灭弧等因素的多维度制约,目前,尚未发现综合性能全面优于SF6的单质绝缘气体[4~6].
三氟甲基磺酰氟(CF3SO2F)是一种潜在的SF6替代气体.得益于>SO2基团的反应活性,化工行业中CF3SO2F常用于制备各种含氟药物、催化剂的原料及中间体.在高压电气领域,CF3SO2F最早用于卤代烷烃绝缘气体的添加剂,如向CCl2F2绝缘气体中添加25%CF3SO2F则可以抑制碳化现象,有效消除碳沉积问题[7].早在1982年,Wootton等[8]研究了CF3SO2F的绝缘特性,发现官能团CF3SO2—的吸电子能力比—C≡N基团更强,CF3SO2F气体的绝缘强度约为SF6的1.4倍,且气体毒性远低于含—C≡N的有机腈化合物.更重要的是,CF3SO2F的正常沸点为-21.7℃,与C4F7N气体(-4.7℃)相比,显然具有更优的低温应用环境适应能力.同时,Wootton等[8]发现体积比为1∶1的CF3SO2F/SF6混合气体的灭弧特性可达到纯SF6的70%~80%.最近,CF3SO2F作为SF6替代气体引起了广泛关注,并对其绝缘性能进行了系统的理论与实验研究[9~12].
稳态汤生(SST)实验表明,CF3SO2F气体的电离系数(α)比SF6低,但电子附着系数(η)高于SF6,临界场强(E/N)lim为5.788×10-15V·cm2,约为SF6的1.6倍.针对混合气体应用方案,体积混合比为1∶4的CF3SO2F/N2气体的交流击穿场强能够达到SF6的80%;当混合比提高至2∶3时,CF3SO2F/N2气体的绝缘性能则明显优于SF6,而此时混合气体的GWP仅为SF6的10%,环保收益非常显著.
将CF3SO2F用于替代高压电气设备中的SF6气体、开展绝缘设计与放电试验之前,需掌握CF3SO2F绝缘介质的各种理化特性,特别是气-液相平衡、气体密度、热容、临界点、介电常数、黏度、扩散系数及热导率等基础关键参数.但是,至今鲜有关于CF3SO2F分子理化性质的理论或实验研究的报道.1956年,Gramstad和Haszeldine[13]以CH3SO2F或CH3SO2Cl为 原 料,采 用 电 化 学氟 化 方 法 合 成 了CF3SO2F,并测量了226~249 K温度范围内的蒸气压,获得标准大气压下CF3SO2F的正常沸点为-21.7℃,蒸发潜热为23.4 kJ/mol.1980年,Katsuhara等[14]测量了低温液态CF3SO2F的拉曼光谱.2000年,Haist等[15]采用气相电子衍射技术测量了CF3SO2F的分子结构参数.本文采用分子动力学(MD)模拟方法,基于量子化学计算,建立了适用于CF3SO2F体系的力场模型,首次预测了CF3SO2F绝缘介质理化特性的各种基本参数,为推动CF3SO2F替代SF6的研究提供了理论依据.
1 计算方法
为了正确描述CF3SO2F分子体系的理化特性,首先采用现有的、且已被广泛认可的力场模型[如CVFF(Consistent-valence forcefield),PCFF(Polymer-consistent forcefield),COMPASS(Condensed-phase optimized molecular potentials for atomistic simulation studies)等]尝试优化CF3SO2F的分子结构[16],并计算了其振动频率与偶极矩.但这些成熟的力场均给出不合理甚至错误的CF3SO2F分子结构参数及性质,显然并不适用于模拟CF3SO2F体系.因此,采用量子化学计算方法,在M06-2X/Aug-cc-pVT(+d)Z理论水平[17](M06-2X/AVTZ+d)优化CF3SO2F分子结构及其构象转变能量途径.在同样理论水平下进行非谐性振动分析,获得分子的振动频率,计算其电偶极矩,并以此为基础重新优化力场势能参数,建立适用于CF3SO2F体系的新力场模型.
为了模拟CF3SO2F的气-液共存相平衡,原则上可以采用Gibbs巨正则系综Monte-Carlo(MC)方法模拟蒸发与凝聚过程,但此方法非常耗时且难以达到真正的平衡态.本文设计了一种更为可靠的简便模拟方法:首先建立由256个CF3SO2F分子随机组成的液体盒子模型,初始密度设置为1.0 g/cm3.基于实验测量的蒸气压-温度关系[13],确定温度T(K)下饱和蒸气压p(MPa),应用周期性边界条件,以恒温恒压(NPT)系综模拟CF3SO2F液体:

采用Nose-Hoover控温和Brendsen控压方法,时间步长设为1 fs,经5 ns的动力学平衡后,继续模拟10 ns用于分析温度T对应的液体密度(ρliq,g/cm3).实际气体密度(ρvap,g/cm3)采用温度依赖的二级Virial系数B2(cm3/mol)校正获得:

式中:Uinter(kJ/mol)为任意随机取向的两个CF3SO2F分子之间的相互作用能;r12(nm)为分子间质心距离;R(8.314 J·k-1·mol-1)为气体常数.
随机选取约4百万个CF3SO2F分子对进行系综平均,采用数值积分方法计算B2.基于校正后的ρvap,建立由256个CF3SO2F分子组成的周期性盒子,采用恒温恒容(NVT)系综模拟CF3SO2F气体的理化特性.与传统分子蒙特卡罗(MC)或MD相平衡模拟方法相比,将气相与液相分开进行模拟,不仅确保气液两相始终处于真正的平衡态,而且显著提高了计算效率.显然,此方法不能模拟气液相界面性质,无法获得表面张力等参数.虽然界面是气液相平衡的重要组成部分,但对于达到平衡后气体或液体的均相密度并没有影响.另外,虽然Gibbs系综方法也可以获得相结构与热力学参数,但计算分子输运特性仍以MD模拟方法更为有效.为了考察可能的尺寸效应,采用包含1024个CF3SO2F分子体系进行了验证计算.与256个分子体系相比较,相同温度下CF3SO2F液体密度的变化普遍小于1%,在MD统计误差范围内.因此,为了兼顾计算精度与效率,在实际计算中均采用256个分子并应用周期性边界条件模拟CF3SO2F气体与液体.
根据不同温度下气-液相平衡的密度数据,CF3SO2F的临界温度(Tc,K)可采用Ising模型拟合获得:

式中:B为待定常数,β=0.325.同理,临界密度(ρc,g/cm3)由下式计算:

式中:A为待定常数.与饱和蒸汽压直接相关的蒸发焓(ΔHvap,kJ/mol),可根据所模拟CF3SO2F液体的能量参数计算:

式中:Eliq(T)(kJ/mol)为温度T下所模拟液体的分子间相互作用能;pΔV(kJ/mol)为蒸发膨胀功;ΔHreal-ideal(kJ/mol)为实际气体与理想气体焓的差,通常可以忽略不计.
CF3SO2F的扩散系数D(m2/s)由分子质心的均方根位移计算得到:

式中:r(t)为t时刻分子质心运动的位置向量;t(s)为分子质心运动的时间.
为了确保计算结果的可靠性,采用两种完全独立的理论方法计算CF3SO2F液体与气体的介电常数.方法1是基于动力学平衡态的Kirkwood-Frohlich方程:

式中:εRF为连续介质反应场参数;M为体系的总偶极矩;V(m3)为系统体积;kB为Boltzmann常数;ε0为真空介电常数.
方法2是引入外加电场进行非平衡动力学模拟[18]:

式中:Ezext为沿模拟体系z轴方向施加的电场强度;Mz为体系沿z轴方向的偶极矩.外加电场强度设置在0~108V/m范围内,确保体系始终处于线性响应区间.
黏度(η,Pa·s)采用体系压力自相关函数计算,即Green-Kubo公式:

式中:Pαβ(t)为t时刻体系压力张量非对角元.
采用非平衡动力学方法计算热导率(λ,W·m-1·K-1)[19].首先,将包含256个CF3SO2F分子的立方体盒子沿z轴方向扩展,形成边长Lx=Ly=Lz/4的正交盒子,共包含1024个CF3SO2F分子,并将盒子沿z轴方向划分为40个温度区间;然后将设定能量(jz,GW/m2)以微扰项引入模拟体系,驱动体系能量从低温区向高温区流动;当达到稳态时,单位时间、单位面积上同样的能量jz沿相反方向自然流动(即热传导),最终实现能量守恒并获得相应的温度梯度(dT/dz),热导率为

应用周期性边界条件,基于NVT系综,设置时间步长为1 fs,每隔100 ps交换各区间能量(原子速度),共进行10 ns动力学模拟,取最后5 ns轨迹分析热导率.
2 结果与讨论
2.1 mPCFF力场模型
CF3SO2F分子结构可以看作由CF4与SO2化学键杂化而形成[9],具有平面对称性,属于Cs点群,其中二面角F—C—S—F呈反式,最稳定构象呈交叉式构型,重叠式构象则为CF3绕C—S键内转动的鞍点(图1).分别采用CVFF,PCFF和COMPASS力场优化CF3SO2F,所得结构参数列于表1.可见,CVFF力场预测—SO2基团结构与实验值偏差显著,S=O键偏长而O=S=O键角偏小,S—F键偏长,F—S—C键角偏大;PCFF结果比CVFF稍有提高,但其中S—F键与F—S—C键角仍偏大;COMPASS力场计算的S—F键长、F—S—C键角均与实验值接近,但给出了错误的F—C—S—F二面角,即COMPASS预测重叠式构象为最稳定结构,与实验测量结果严重不符.量子化学方法M06-2X/AVTZ+d优化得到的CF3SO2F结构参数与实验值吻合得很好,预测交叉式比重叠式构象的能量低11.3 kJ/mol,分子偶极矩为4.84×10-30C·m.为了考察计算方法对分子结构参数的影响,采用更大的aug-cc-pVQZ+d(简记为AVQZ+d)基组,重新优化了CF3SO2F结构参数(表1),发现键长、键角、内转动势垒及偶极矩等参数基本保持不变.

Fig.1 Molecular structure of CF3SO2F

Table 1 Predicted parameters for CF3SO2F at various levels of theory
显然,CVFF,PCFF,COMPASS计算的分子结构参数、稳定构象、CF3内转动势垒及偶极矩等均与实验或量子化学计算结果偏差较大,特别是CVFF和COMPASS力场,无法直接用于CF3SO2F分子理化性质的分子动力学模拟.
选取与M06-2X/AVTZ+d计算结果相近的PCFF力场进行改造,重新优化CF3SO2F的力场参数,主要包括3个键长(C—S,F—S,O=S)、2个键角(C—S—F,O=S=O)以及1个二面角(F—C—S—F)所需的势参数.采用最小二乘法,拟合M06-2X/AVTZ+d的结构参数、振动频率、内转动势能曲线,建立了mPCFF新力场,并与原始PCFF力场参数进行对比(表2).
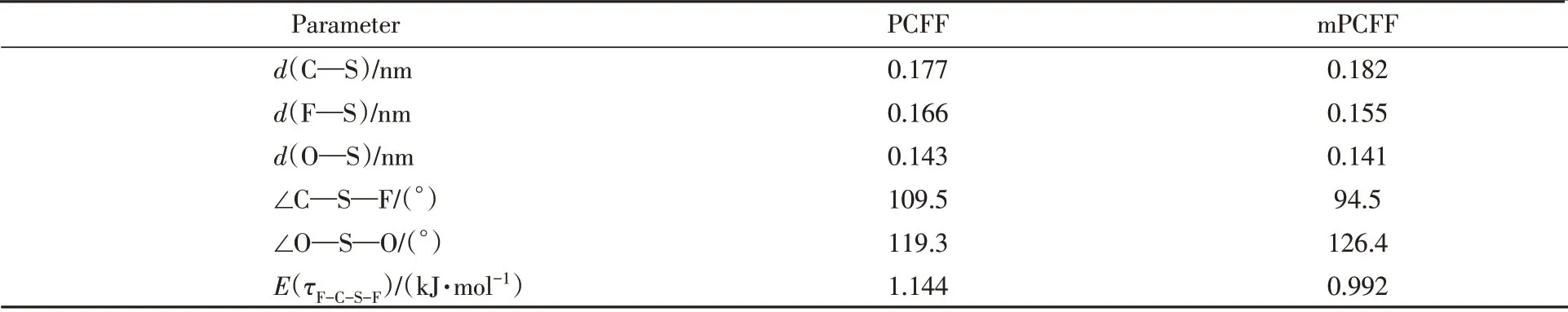
Table 2 Parameters for PCFF and mPCFF force fields
从表1可见,mPCFF优化的CF3SO2F结构参数与实验结果[15]一致,相对误差仅为0.3%;mPCFF计算的内转动势能曲线与M06-2X/AVTZ+d结果相吻合(图2),较原始PCFF结果有明显改进.同时,mPCFF力场计算的振动频率与M6-2X/AVTZ+d和实验结果[14]也较为一致(图3).mPCFF力场预测CF3SO2F分子的偶极矩为4.47×10-30C·m,与M06-2X/AVTZ+d的计算结果(4.84×10-30C·m)相符,较原始PCFF的3.64×10-30C·m有显著提高.总之,优化的mPCFF力场模型能够正确描述CF3SO2F的结构与性质(见本文支持信息).

Fig.2 Profiles for the internal rotation of CF3 around the C—S bond of CF3SO2F calculated at various levels of theoryThe insets show the conformations with dihedral angles(F—C—S—F)of 60°and 120°,respectively.
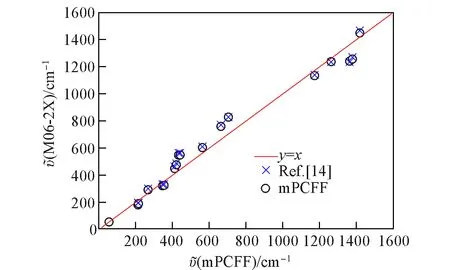
Fig.3 Vibrational frequencies of CF3SO2F calculated by mPCFF force field compared with the experimental data and correlated to the M06-2X/AVTZ+d theoretical data
值得指出的是,改进的mPCFF力场不仅能够正确描述CF3SO2F分子的结构和性质,还适用于描述其它包含磺酰氟基团的同类分子体系(如SO2F2分子),mPCFF预测的偶极矩为3.54×10-30C·m,与实验测量结果[(3.54±0.07)×10-30C·m]和M06-2X/AVTZ+d计算值(3.84×10-30C·m)均相吻合.
化合物的热力学和输运性质强烈依赖于非键相互作用参数(包括范德华力、静电力等),PCFF力场中的非键相互作用参数相当成熟,能够成功描述很多有机、无机化合物的热力学性质.为了考察非键相互作用力场参数的可靠性,采用M06-2X/AVQZ+d方法优化获得了3种代表性的(CF3SO2F)2二聚体结构D1,D2和D3,并依此计算了分子间相互作用势能随S—S,S—C和C—C原子间距的变化,如图4(A)~(C)所示.基于量子化学计算的分子结构参数,即采用mPCFF力场计算的分子间势能与M06-2X/AVQZ+d的计算结果一致.显然,mPCFF力场能够合理描述CF3SO2F的分子间相互作用.
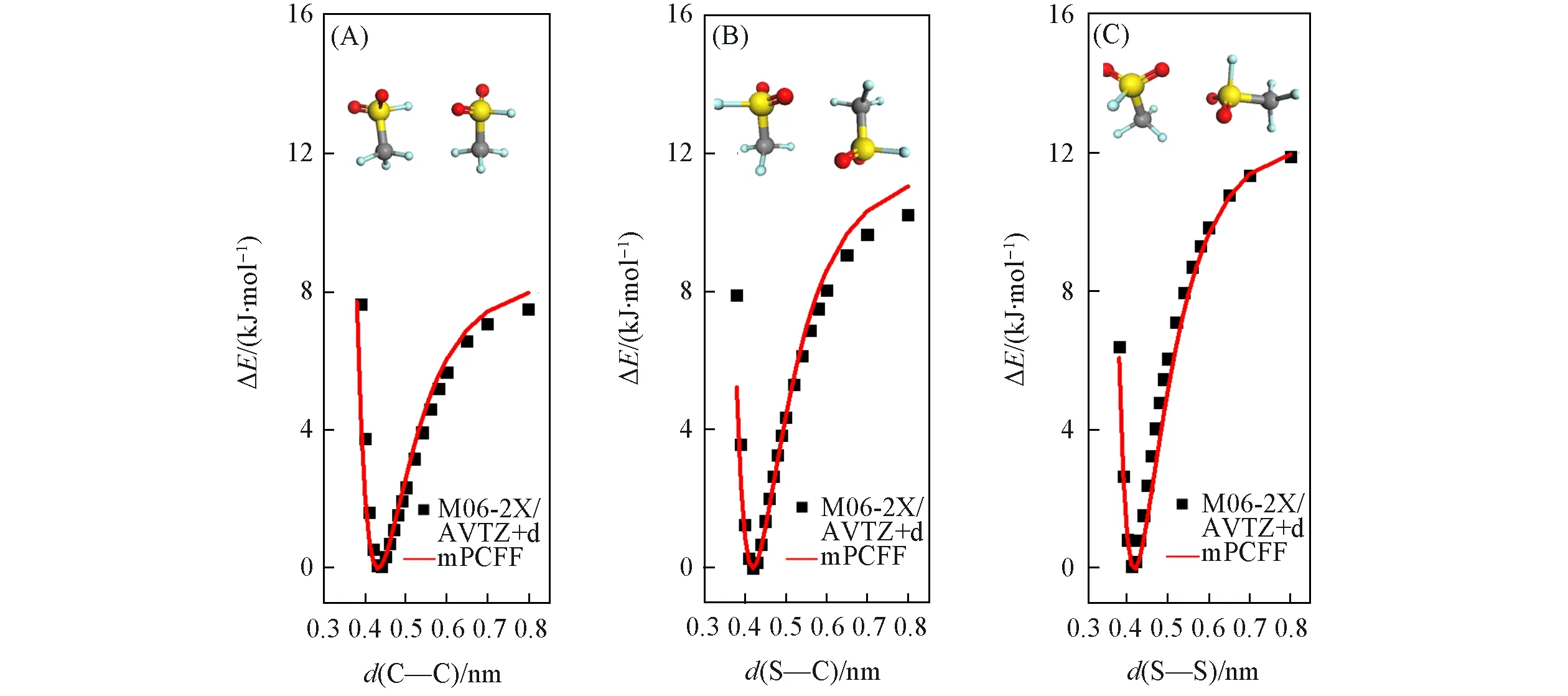
Fig.4 Profiles for the intermolecular interactions between two CF3SO2F molecules at three different dimeric conformations of D1(A),D2(B)and D3(C)The insets show the three corresponding conformations.
2.2 气-液相平衡
基于mPCFF力场,采用分子动力学方法模拟了243~323 K温度范围CF3SO2F的气-液相平衡性质,结果列于表3.

Table 3 Predicted vapor pressures,virial coefficients,densities,and heat capacities of CF3SO2F
CF3SO2F液体的饱和蒸汽压随温度的变化与蒸发焓有关,可用Clausius-Clapeyron方程表示:
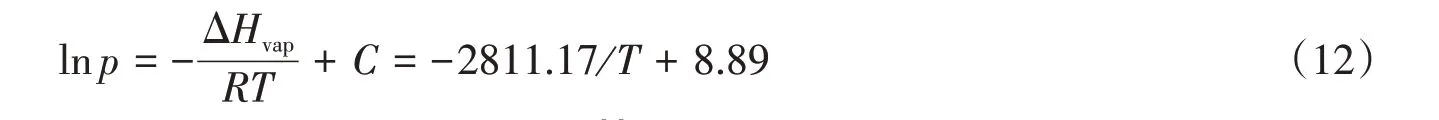
式中:ΔHvap(kJ/mol)为蒸发焓;C为常数.由式(12)得到CF3SO2F的ΔHvap=23.4 kJ/mol.另一方面,基于CF3SO2F液体的动力学模拟轨迹也可直接计算温度依赖的蒸发焓,结果如图5所示.可见,蒸发焓随温度升高呈线性下降,可表示为ΔHvap=41.7-0.0623T.由此计算室温(298.15 K)下蒸发焓ΔHvap=23.1 kJ/mol,与实验结果相吻合,表明mPCFF力场描述CF3SO2F气-液相平衡较为可靠.各温度下CF3SO2F气相热容(Cv)列于表3,由于NVT系综下饱和蒸气的浓度差异,Cv随温度的变化较为复杂,在0.2~1.3 kJ·mol-1·K-1范围内呈现起伏.

Fig.5 Theoretical temperature-dependent heats of vaporization of CF3SO2F compared with the experimental valueDashed line:linear fit to the theoretical data.
根据模拟得到的气-液相平衡密度数据,预测CF3SO2F的临界温度Tc=(453±2)K,临界密度ρc=(0.8126±0.0127)g/cm3.与C4相比[20],CF3SO2F的沸点降低了约17 K,但其Tc比C4的Tc提高约76 K,ρc比C4的ρc增加0.26 g/cm3,表明CF3SO2F比C4具有更宽的相平衡区间,比C4具有更优的环境温度适应性.
CF3SO2F气体密度与饱和蒸汽压之间可通过二级Virial系数关联:

值得指出的是,在243~323 K温度范围内,模拟发现饱和蒸汽压与气体密度基本呈简单线性相关:

表明CF3SO2F分子间相互作用较弱,在所考察的温度范围内接近于理想气体,为工程应用快速估算电气设备中CF3SO2F气体的密度和饱和压力提供了理论依据.
2.3 输运动力学性质
CF3SO2F在液相与气相中的扩散系数随温度的变化关系如图6所示.显然,在压缩液体中CF3SO2F的扩散系数较低,随温度的升高而增加,扩散符合Arrhenius行为,可拟合为

由此得到CF3SO2F液体的自扩散活化能为10.1 kJ/mol.与此相反,在气相中CF3SO2F的扩散系数(图6)随温度升高而下降,呈负温度效应,这是由于处于饱和蒸汽压下的气体密度随温度的升高而迅速增加(表3),从而导致分子自扩散减慢.采用Arrhenius公式拟合气相扩散系数,可得:

为了考察体积固定的电气设备中CF3SO2F气体的扩散行为,取室温(298.15 K)下饱和蒸汽所对应的气体密度ρvap=0.0404 g/cm3,基于NVT系综模拟了243~323 K温度范围内CF3SO2F气体扩散系数,结果如图6所示.可见,CF3SO2F气体的扩散系数随温度的升高而增加,通过Arrhenius公式拟合得到气相扩散活化能为4.8 kJ/mol,约为液相活化能的50%,表明CF3SO2F虽然在气相中扩散比液相快约1个数量级,但受温度的影响较弱,相对而言,液体中CF3SO2F分子扩散动力学特性随温度变化更为敏感.
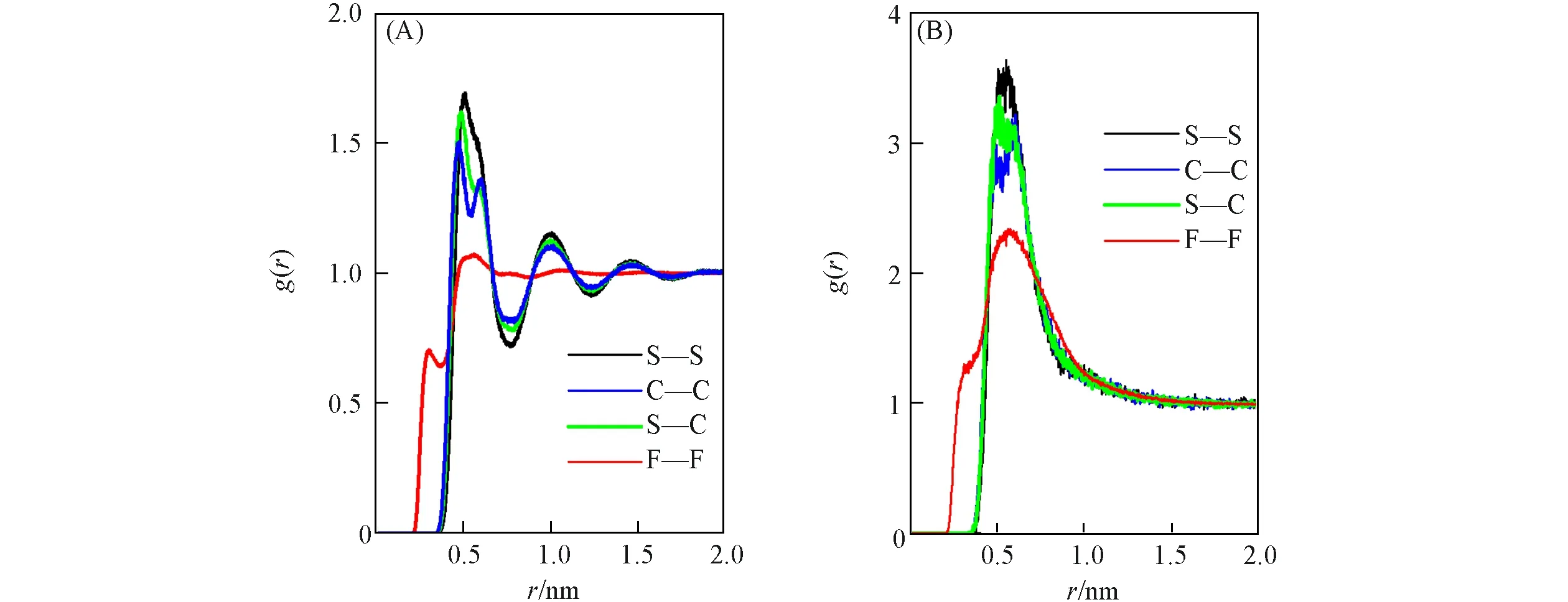
Fig.7 Radial distribution functions of CF3SO2F in liquid(A)and vapor(B)phases
CF3SO2F的自扩散行为与其在液相或气相中的结构有密切关系.图7(A)和(B)分别给出了298.15 K压缩液体(p=0.583 MPa)及饱和气相中CF3SO2F各原子间的径向分布函数.可见,CF3SO2F分子间相互作用较弱,只有液相中的径向分布函数存在微弱F—F峰(约0.3 nm),强度仅为0.7,气相中F—F原子间作用峰则不明显.其余—SO2,—CF3基团之间的相互作用峰,即S—S,C—C和S—C,均出现在0.4~0.6 nm之间,表明CF3SO2F分子间作用将以长程势(如静电势)为主,短程范德华作用贡献较小,与CF3SO2F具有较低的沸点一致.从径向分布函数峰所对应强度分析,推测在气相或液相中—SO2基团之间的相互作用比—CF3基团稍强,前者将是决定CF3SO2F理化特性的关键结构因素.
CF3SO2F液体与气体在243~323 K温度范围内的静态介电常数如图8所示.值得指出的是,依据方法1和方法2计算得到的液体介电常数相当一致,基本不受理论方法的影响,表明MD模拟所得介电常数自洽可靠.
从具体数据来看,方法2计算得到的介电常数随温度的振荡起伏稍大,但总体上仍保持线性趋势不变.对于CF3SO2F液体,其介电常数从243 K时的3.5降为323 K时的2.6.虽然CF3SO2F分子的极性较强,其偶极矩高达H2O分子的80%,但其液相的介电常数较小,仅为液体水的4%左右,表明介电常数不仅与偶极矩有关,而且受制于分子结构及分子间相互作用模式.CF3SO2F液体的介电常数随温度的变化关系可表示为

相比较而言,CF3SO2F气体的介电常数随温度的升高而快速增加,但在所考察的温度范围内基本处于1.0~1.1范围.
模拟发现CF3SO2F液体在253.15 K(接近沸点温度)时的黏度为1.44×10-3Pa·s,随着温度升高,其黏度逐渐降低,293.15 K时压缩液体的黏度仅为5.10×10-4Pa·s.相应地,CF3SO2F气体黏度远低于液体黏度,随温度的变化更为敏感,且呈现正温度效应,从253.15 K时的3.4×10-7Pa·s增加到293.15 K时的1.00×10-4Pa·s.

Fig.8 Static dielectric constants of CF3SO2F in the vapor-liquid equilibria

Fig.9 Temporal profiles for temperature gradient,energy flux,and thermal conductivity of CF3SO2F at 293.15 K
以温度293.15 K时CF3SO2F气体的热传导为例,图9给出了典型的温度梯度、能流密度以及热导率模拟计算结果.可以看出,随着能量在高温和低温区间的反复交换,整个气相体系在约5 ns模拟时间后基本达到平衡态.模拟发现CF3SO2F气体的热导率受温度的影响较小,在253~323 K范围内计算得到气体热导率的平均值为(0.0092±0.0025)W·m-1·K-1.对于CF3SO2F液体,预测293.15 K下的热导率为0.036 W·m-1·K-1,约为气体热导率的3倍.
2.4 混合气体
为了满足高压电气设备的绝缘需求,气体压力通常达到0.5 MPa或以上.虽然CF3SO2F气体的液化温度较低(-21.7℃),仍需使用低沸点缓冲气体CO2和N2等配制混合气体,才能有效避免低温应用环境中出现液化.原则上可以直接模拟混合气体的液化温度,但因气体单质的液化特性差异显著,达到混合气体的气液相平衡需要长时间的分子动力学模拟或Gibbs系综海量采样,目前仍难以实现.考虑到缓冲气体的液化温度远低于CF3SO2F,为了方便起见,根据CF3SO2F饱和蒸汽压的Clausius-Clapeyron方程,总压力为p时,CF3SO2F体积占比x%的CF3SO2F/N2或CF3SO2F/CO2混合气体的液化温度(Tb)可近似为

如,当总压p=0.7 MPa时,混合气体中CF3SO2F的最高占比分别为19.3%,15.6%,12.5%和9.88%时,所对应的最低使用温度分别为-15,-20,-25和-30℃;当混合比达到50%时,总压0.5和0.7 MPa下所对应的混合气体的液化温度分别为0.4和9.7℃.
在总压力p=0.5 MPa、温度T=298.15 K条件下,基于mPCFF力场分别模拟了混合比为50%的CF3SO2F/N2和CF3SO2F/CO2两种混合气体的各种理化特性参数(表4),并与SF6以及最新的g3混合环保绝缘气体(9%C4+91%CO2)进行对比.可以看出,同等条件下,CF3SO2F与N2混合气体的黏度与扩散系数高于其与CO2的混合气体,因为N2的扩散系数约为CO2的2倍.导致此现象的原因可能与CF3SO2F与N2和CO2分子间相互作用的差异有关,也可能与N2和CO2分子本身的扩散特性有关,同等条件下N2比CO2扩散更快.然而,CF3SO2F/CO2混合气体的热导率比CF3SO2F/N2高约15%.与g3气体相比,CF3SO2F/CO2混合气体的密度、介电常数和黏度均有所增加,热导率相似,而扩散系数则显著降低,因CO2分子在CF3SO2F/CO2混合气体中的扩散系数仅为g3中的3/4,表明CF3SO2F与CO2分子间作用较强.与SF6相比,所有混合气体的热导率均不及SF6,扩散系数亦显著低于SF6.综上所述,当使用CF3SO2F混合气体替代SF6时,对设备中气体的扩散以及热传导特性应给予重点关注.
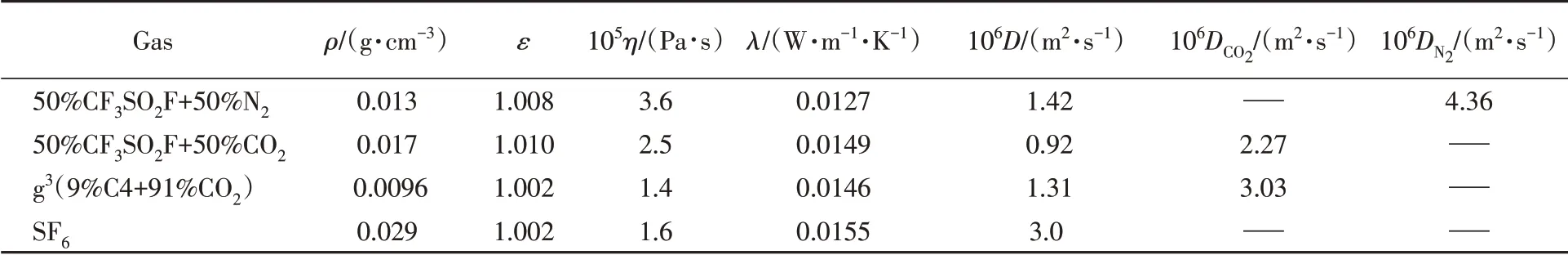
Table 4 Predicted physicochemical properties of the gas mixtures of CF3SO2F with N2 or CO2 at 0.5 MPa and 298.15 K
3 结论
建立了适用于CF3SO2F体系的mPCFF力场模型,采用分子动力学模拟方法,系统研究了CF3SO2F的气-液相平衡及各种输运特性,获得了243~323 K温度范围内CF3SO2F的气液相平衡数据、临界参数、自扩散系数、介电常数、黏度、热导率及其随温度的变化关系.mPCFF力场优于CVFF和COMPASS传统力场,分子模拟结果与实验或量子化学计算结果吻合.CF3SO2F以分子间长程静电作用为主,气体扩散的活化能为4.8 kJ/mol,且饱和蒸汽压与气体密度呈简单线性关系;CF3SO2F气体的介电常数约为1.0~1.1,黏度随温度变化较为敏感,而热导率基本与温度无关.当电气设备充气压力为0.7 MPa时,使用体积占比不超过10%的CF3SO2F与N2或CO2形成混合气体,即可满足最低环境使用温度达-30℃的需求.当CF3SO2F体积占比提高至50%时,混合气体的理化特性与g3气体相近,但气体热导率和扩散系数分别约为SF6的80%和47%.模拟结果可为CF3SO2F替代气体的绝缘设计提供理论参考.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220424.
猜你喜欢
传染病信息(2022年6期)2023-01-12
中国音乐学(2022年1期)2022-05-05
教学与管理(理论版)(2017年7期)2017-08-11
中学化学(2017年5期)2017-07-07
未来英才(2016年13期)2017-01-13
中国塑料(2016年8期)2016-06-27
磁共振成像(2015年9期)2015-12-26
磁共振成像(2015年7期)2015-12-23
肿瘤影像学(2015年3期)2015-12-09
中学理科·综合版(2008年4期)2008-07-15
