
建立具有区分力溶出介质 提升仿制药非布司他的生物等效性
2022-11-17 00:55唐建飞谢红瑜赵福斌郭艳超
药学与临床研究 2022年5期
唐建飞,谢红瑜,赵福斌,方 霞,郭艳超
杭州朱养心药业有限公司,杭州 310018
一致性评价是指对已经批准上市的仿制药,需在质量、疗效、安全性等方面达到与原研药一致的水平,实现临床上替代原研药的目的[1]。开展仿制药一致性评价,有助于提高药品的有效性和安全性,为临床用药提供更多选择,也降低了患者治疗费用与节省国家医保支出。
痛风以及经常发作痛风性关节炎、慢性痛风性关节病、痛风性肾病和尿酸性结石病的长期治疗需使用降尿酸药物,其主要作用途径为抑制尿酸生成和促进尿酸排泄。而抑制尿酸生成的常用药物为别嘌醇和非布司他[2,3]。非布司他最先由日本Teijin Pharma研制开发,于2008 年10 月被欧洲EMA 批准上市,商品名为Adenuric®;2009 年2 月经美国FDA 批准上市,商品名为Uloric®[4]。2012 年美国风湿病学会在痛风指南中将非布司他推荐为高尿酸血症、痛风的A 级治疗药物。
非布司他为选择性高、效果显著的黄嘌呤氧化酶抑制剂,通过作用于黄嘌呤氧化酶的钼蝶呤中心,使氧化态或还原态的钼辅因子保持孤立状态,从而减少黄嘌呤氧化酶与底物的结合,减少尿酸的生成,降低血液中尿酸的浓度,从而达到治疗高尿酸血症或痛风的作用。
本研究重点论述仿制药非布司他片的开发及生物等效性评价问题。
1 仪器与药品、试剂
仪器设备:708DS 全自动溶出仪(安捷伦公司);Angilent G6460 型三重四级杆液质联用仪;紫外-可见分光光度计(安捷伦公司);XS205 万分之一天平(梅特勒公司);pH计。
药品:A 制剂非布司他片(风定宁®,批号L190101,40 mg/片)、B 制剂非布司他片(风定宁®,批号L190201,40 mg/片)、C 制剂非布司他片原研药(Uloric®,批号A26679,40 mg/片)。
试剂:十二烷基硫酸钠(SDS,含量99%,西格玛公司,含量>85%,西陇化工)、磷酸二氢钾、氢氧化钠、醋酸钠、盐酸均为分析纯;纯化水(自制)。
2 方法与结果
2.1 体外溶出试验
为确保仿制制剂与参比制剂的体外溶出对比方法准确可靠,保证结果的可靠性,需要对溶出曲线检测方法进行方法学验证。
选择验证的介质4 种:0.5%SDS 溶液、pH 1.2(含0.5% SDS)、pH 4.5(含0.5% SDS)和pH 6.8 磷酸盐缓冲液。验证项目包括专属性、滤膜吸附、溶液稳定性、准确度、精密度、线性与范围。验证结果:专属性良好,空白辅料和空白溶剂对检测无影响;滤膜吸附试验结果表明,当弃去4 mL 初滤液时达到吸附饱和;对照品溶液和供试品溶液在室温条件下6 h 内稳定性良好;在0.5% SDS 介质中平均回收率为100.3%,在pH 1.2 的0.5% SDS 中平均回收率为97.2%,pH 4.5 的0.5% SDS 平均回收率为98.8%;在pH 6.8 磷酸盐缓冲液中的平均回收率为99.7%;在4 种介质中回收率良好;重复性和中间精密度的结果RSD 均<2%;线性在标示量的10%~120%浓度范围内相关系数均>0.999 9。
2.1.1 具有区分力的介质筛选
2.1.1.1 非布司他在不同介质中饱和溶解度 依照缓冲液配制方法(《中国药典》2020 年版四部通则8004),配制pH 1.0、pH 2.2 盐酸溶液,pH 3.8、pH 4.5醋酸盐缓冲液,pH 5.5、pH 6.0、pH 6.8、pH 7.6 磷酸盐缓冲液,以及质量分数分别为0.30%、0.35%、0.40%、0.45%、0.50%的十二烷基硫酸钠(SDS)溶液,取本品在上述介质中进行饱和溶解度试验。结果见表1。

表1 非布司他在不同pH 条件下的溶解度
由表1 可知,本品具有pH 值依赖性,这与日本IF 文件数值一致[5];随着SDS 浓度增加,非布司他溶解度增加,因不同厂家的SDS 纯度不同,pH 值会有差异。本次研究使用的SDS 采用西格玛品牌,纯度大于99%,减少了其纯度对结果的影响。根据原料在不同浓度SDS 中的溶解度,将进一步考察原研制剂在不同浓度pH 值介质中的溶出曲线。
2.1.1.2 原研制剂在不同pH 值介质中的溶出曲线 对同一批原研样品在不同pH 值介质中进行曲线考察,结果见图1。

图1 原研品不同pH 值介质中溶出曲线
从曲线图中可知,制剂在pH 1.2 和pH 4.5 介质中溶出缓慢,在pH 6.8 和pH 6.0 介质中溶出较快,均无法达到仿制品与原研品有效对比的目的;在pH 5.5 介质中溶出适中,但考虑到本品对pH 值依赖性明显,对溶出介质的配制误差接受度较低,故从不同浓度SDS 水溶液中对原研品溶出曲线的影响进行筛选。
2.1.1.3 原研制剂在不同浓度SDS 介质中的溶出曲线 在不同pH 值缓冲液中加入SDS 不会影响pH值,多次配制的介质pH 结果见表2;不同品牌的SDS 水溶液的pH 值差异较大,但将纯度较大的同品牌SDS 将不同时间配制并加入不同浓度的SDS水溶液时,pH 值未见明显差异。SDS 水溶液其pH值结果见表3;本次研究选择纯度较高的SDS(西格玛)水溶液进行曲线条件筛选,结果见图2。

表2 不同pH 介质中加入不同浓度SDS 的pH 值测定结果

表3 不同浓度SDS 水溶液的pH 值测定结果
从图2 曲线可知,0.3% SDS 溶出曲线较低,不利于观察仿制品与原研品溶出曲线是否一致,故初步选择0.5% SDS 作为关键介质。

图2 原研品不同浓度SDS 介质中溶出度曲线
2.1.1.4 区分不同工艺样品溶出介质的筛选 取处方组成一致、工艺中采用不同工艺参数制备2 个制剂,用A 和B 表示。
A 制剂制粒搅拌速度为360 r·min-1,B 制剂制粒搅拌速度为180 r·min-1。取两种制剂各12 片,依照溶出度与释放度测定法(《中国药典》2020 年版四部通则0931 第二法),分别以pH 1.2 的0.5% SDS、pH 4.5 的0.5% SDS、pH 6.8 磷酸盐缓冲液和0.5%SDS 水溶液各900 mL 作为溶出介质,设置温度为37℃,转速为75 r·min-1,于5、10、15、30、45、60 min 自动取样5 mL,滤过,精密量取续滤液2 mL,置10 mL量瓶中,加溶出介质稀释至刻度,摇匀,即为供试品溶液。
精密称取非布司他对照品10 mg,置25 mL 量瓶中,加乙腈-水(1∶1)适量,超声溶解并稀释至刻度,摇匀,精密移取1 mL 置50 mL 量瓶中,用溶出介质稀释至刻度,摇匀,作为对照品溶液。
取上述供试品溶液和对照品溶液,按照紫外-可见分光光度法(《中国药典》2020 年版四部通则0401),于315 nm 波长处测定吸光度,计算不同条件下非布司他累积溶出度,结果见图3~图6。

图3 A 制剂和B 制剂在pH 1.2 的0.5% SDS 介质中曲线比较

图4 A 制剂和B 制剂在pH 4.5 的0.5% SDS 介质中曲线比较

图5 A 制剂和B 制剂在pH 6.8 磷酸盐溶液中的曲线比较
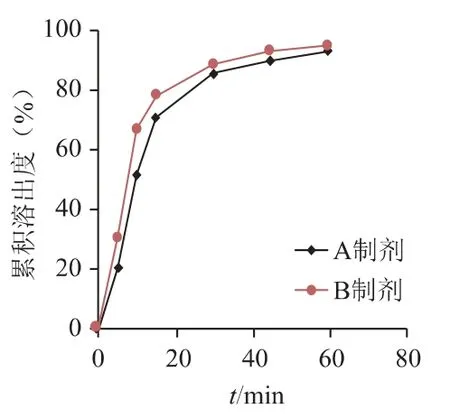
图6 A 制剂和B 制剂在0.5%SDS 介质中曲线比较
差异因子f1是衡量两条曲线相对偏差的参数,f1越大,说明两条溶出曲线的差异越显著。因此,可以通过差异因子(f1)法计算图3~6 中两条溶出曲线的总体差异,计算公式如下:

其中n 为取样时间点个数,Rt为A 制剂在t 时刻的累积溶出百分率,Tt为B 制剂在t 时刻的累积溶出百分率,计算结果见表4。

表4 A 制剂和B 制剂在不同介质中差异因子f1
从图7 可知3 批原研品在0.5% SDS 介质中无明显差异;但从图6 和表2 可知,不同工艺的仿制品在0.5% SDS 水溶液中溶出具有一定的差异性,故选择0.5% SDS 作为关键介质,筛选处方工艺。

图7 不同批次原研品在0.5% SDS 介质中溶出曲线
2.1.2 自制片与原研[1]片溶出比较 以“2.1.1.4”方法,测定仿制片A、B 与原研制剂C 在0.5% SDS 介质中溶出曲线,结果见表5。
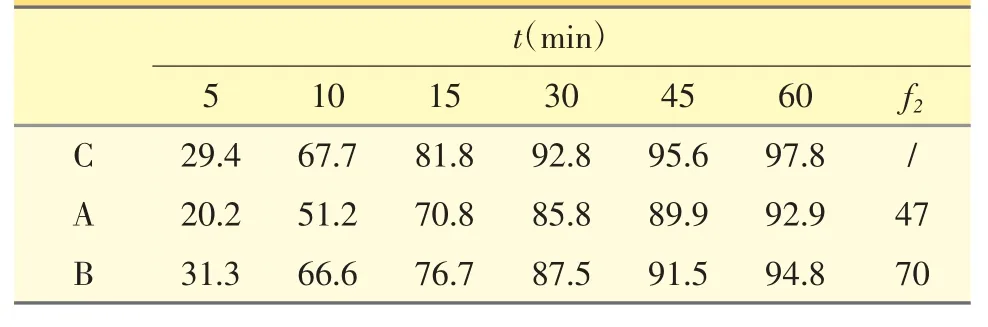
表5 仿制片与原研片在0.5% SDS 介质中药物释放曲线比较
相似因子f2是衡量两条曲线相似程度的参数,f2越大说明两条曲线相似性越高。从表5 可知,A 制剂和原研制剂的相似因子f2小于50,而B 制剂相似因子远大于50,说明B 制剂和原研制剂曲线相似性较高,在体内更易达到生物等效。
2.2 体内试验
采用仿制片A 和B 及原研制剂C1 进行体内生物等效性试验。
试验单位:海口市人民医院,
伦理审核单位:海口市人民医院生物医学伦理委员会
2.2.1 受试者选择 根据临床研究方案,招募24位健康受试者,男女比例适宜,将受试者随机均分为两个试验组(T1 组和T2 组)。所有入组的受试者均符合试验方案的入选标准。
2.2.2 分组给药与血样本采集 本试验采用随机、开放、两周期、自身交叉试验设计,洗脱期为7 天。将24 位健康受试者按1∶1 随机分为T1、T2 试验组,两组男女比例相同。
T1 试验组:将12 位受试者按1∶1 随机分TR、RT 两个给药组:第一周期给药当天按药物随机表,12 位受试者用240 mL 温水送服1 片参比制剂(R、商品名:Uloric®,40 mg/片)或1 片受试制剂A(TA、40 mg/片)。洗脱期7 天,第二周期按药物随机表,12位受试者服用对应的T 或R 制剂。
T2 试验组:将12 位受试者按1:1 随机分TR、RT两个给药组:第一周期给药当天按药物随机表,12 例受试者用240 mL 温水送服1 片参比制剂(R、商品名:Uloric®,40 mg/片)或1 片受试制剂B(T、40 mg/片)。洗脱期7 天,第二周期按药物随机表,12 位受试者服用对应的T 或R 制剂。
餐后试验每周期,分别在给药前(0 h)及给药后0.25、0.5、0.75、1、1.25、1.5、1.75、2、2.5、3、3.5、4、5、6、8、12、24h 共18 个采血点,每次采集受试者上肢静脉血约4mL。采集后的全血置于冰浴的EDTA-K2 采血管中,进行离心处理(2℃~8℃、2000g、10min),离心结束后获得血浆,将血浆分装置于检测管及备份管中(每管血浆不少于0.8mL)。分装结束后,将检测管及备份管存放于超低温冰箱(≤-60℃)中保存待测。
2.2.3 血药浓度测定及安全性
2.2.3.1 血药浓度测定 采用LC-MS/MS 法,将EDTA-K2 抗凝的血浆样品作蛋白沉淀提取后,取上清液加50%乙腈-水稀释进样,以非布司他-d7 为内标,检测人血浆中非布司他浓度。
液相色谱条件:以十八烷基硅烷键合硅胶为固定相(Angilent Poroshell 120EC-C183.0 mm×50 mm,2.7 μm),0.1%甲酸水溶液-乙腈(20∶80)为流动相,柱温25℃,流速0.4 mL·min-1,进样量5 μL,运行时间5 min。
质谱条件:采用ESI 源,正离子MRM 模式检测。
对检测方法进行了验证,内容包括系统适用性、线性与范围、精密度、准确度、提取回收率和稳定性等。验证结果:系统适用性良好,5 次重复分析所得待测物与内标色谱峰面积比值的变异系数小于10%;非布司他标准曲线在5~4000 ng·mL-1范围内r2>0.999 9,线性良好;批内和批间精密度的变异系数均小于10%,说明精密度良好;定量下限(LLOQ)浓度、低浓度、中浓度、高浓度的准确度均在90%~110%,说明准确度良好;待测物提取回收率范围为94.7%~96.5%,变异系数为0.9%,内标物的提取回收率97.6%,变异系数为1.2%,说明待测物和内标物提取回收率均良好;非布司他全血样品在冰水浴中可稳定5 h,血浆样品至少4 个冻融循环(-60℃)稳定,制备后样品在6℃环境中存储70h 稳定,样品处理液6℃条件可保存61h 后再进样,生物样本在-60℃及-20℃条件下可长期存放38 d。结论:经验证本方法可用于测定人血浆中的非布司他浓度。
2.2.3.2 安全性状况 采用非房室模型(NCA)计算非布司他的药动学参数,观察所有受试者在临床研究期间发生的任何不良事件,对不良事件发生率、不良事件与试验药物相关性分析及严重程度分析等进行统计描述。在本次研究过程中,无严重不良事件发生,试验过程中无受试者死亡报告。
2.2.4 统计学分析 分析血药浓度(c)-时间(t)、PK 参数,采用BE 集,将主要药代参数Cmax、AUC0-t、AUC0-∞对数转换后进行方差分析(ANOVA),计算两药主要药代参数的几何平均值比值的90%置信区间。比较试验制剂与参比制剂活性成分的主要药代参数,当受试制剂与参比制剂Cmax、AUC0-t和AUC0-∞最小二乘几何均值比值的90%置信区间在80.0%~125.0%等效范围内,则判断为两制剂生物等效。
2.2.5 体内生物等效性分析
2.2.5.1 餐后血药浓度(c)-时间(t)曲线 A 组受试者口服参比制剂(Reference,R)和受试制剂(Test,TA)后血药浓度(c)-时间(t)曲线,见图8。B 组受试者口服参比制剂(Reference,R)和受试制剂(Test,TB)血药浓度(c)-时间(t)曲线,见图9。

图8 A 组受试者餐后平均血药浓度-时间曲线
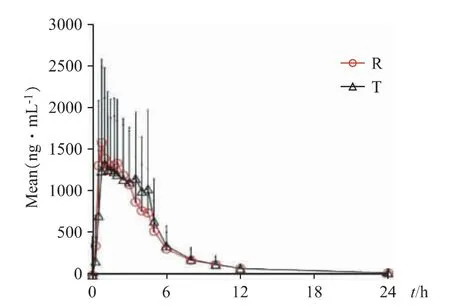
图9 B 组受试者餐后平均血药浓度-时间曲线
2.2.5.2 餐后药代动力学参数 A 组受试者口服非布司他片受试制剂和原研药的主要药代动力学参数见表6、生物等效性评价结果见表7。B 组受试者口服非布司他片受试制剂和原研药:其主要药代动力学参数见表8、生物等效性评价结果见表9。

表6 A 制剂药代动力学参数列表(PKPS)

表7 A 制剂生物等效性评价结果(BES)

表8 B 制剂药代动力学参数列表(PKPS)

表9 B 制剂生物等效性评价结果(BES)
2.2.6 生物等效性评价 经对数转化后双单侧t检验结果表明,A 制剂和原研品非布司他片在餐后条件下Cmax90%置信区间为76.9%~100.0%,AUC0-t90%置信区间为101.5%~105.2%,AUC0-∞90%置信区间为101.5%~105.2%;Cmax未落在80.0%~125.0%等效范围内。A 制剂和原研品非布司他片不具有生物等效性。
经对数转化后双单侧t 检验结果表明,B 制剂和原研品非布司他片在餐后条件下Cmax90%置信区间为95.8%~122.2%,AUC0-t90%置信区间为99.5%~106.7%,AUC0-∞90%置信区间为98.9%~106.0%;均落在80.0%~125.0%等效范围内。B 制剂和原研品非布司他片具有生物等效性。
综上所述,在所选溶出介质+0.5% SDS,75 r·min-1条件下,当A 制剂f2=47(f2<50 溶出行为不一致)时,表现出与原研品体内餐后Cmax不等效,其他指标均等效;当B 制剂f2=70(f2>50 溶出行为一致)时,表现出与原研品体内餐后Cmax、AUC0-t、AUC0-∞均等效。
3 讨论
对于生物药剂学分类系统Ⅱ类(BCS Ⅱ)药物,溶出过程是口服吸收的限速步骤,像非布司他这种难溶且有pH 依赖的BCSII 类品种,在仿制药开发中,达到与原研品体内生物等效难度较高。本研究建立了体内外相关的溶出曲线方法,对指导工艺开发,降低体内不等效的风险具有重要意义。
本研究选择关键工艺参数为变量,制备不同的样品,以0.5% SDS 水溶液为介质进行溶出行为比较,发现A 制剂溶出曲线30 min 前较明显低于原研品,相似因子f2为47,体内餐后Cmax为76.9%~100.0%,下限略低于80.0%,为不等效;B 制剂溶出曲线与原研品非常接近,f2为70,体内餐后Cmax为95.8%~122.2%,为等效。从两组体内外相关数据可知,本研究中的溶出介质可有效区分非布司他片的内在质量,并可通过体外溶出行为预测其体内吸收情况,具有一定的体内外相关性。本研究表明,确保该介质中仿制品与原研品溶出曲线比较f2大于50,可提高体内生物等效性的通过率。
猜你喜欢
内江师范学院学报(2022年4期)2022-04-27
医学概论(2022年4期)2022-04-24
湖北师范大学学报(自然科学版)(2021年3期)2021-09-08
世界最新医学信息文摘(2021年12期)2021-06-09
数学物理学报(2021年1期)2021-03-29
药品评价(2020年13期)2020-10-14
工业设计(2020年6期)2020-07-30
中国生殖健康(2019年2期)2019-08-23
铁道通信信号(2018年9期)2018-11-10
实用妇科内分泌杂志(电子版)(2017年33期)2017-04-01
