
超高效液相色谱-串联质谱法同时测定动物组织中氯霉素、甲砜霉素、氟苯尼考和 其代谢产物氟苯尼考胺残留量
2022-11-21 09:25郑陆红张慧琼黄柳霞
现代食品 2022年20期
◎ 郑陆红,张慧琼,陈 茹,黄柳霞
(1.广东省食品工业研究所有限公司,广东 广州 511442;2.广东省食品工业公共实验室,广东 广州 511442;3.广东省食品质量监督检验站,广东 广州 511442)
氯霉素类药物是一类广谱抑菌抗生素,常见的氯霉素类药物主要包括氯霉素(Chloramphenicol,CAP)、甲砜霉素(Thiamphenicol,TAP)和氟苯尼考(Florfenicol,FF)及其代谢物氟苯尼考胺(Florfenicol Amine,FFA)等[1]。氟苯尼考是一种新型酰胺醇类抗生素,氟苯尼考胺是氟苯尼考的主要代谢产物,通常以氟苯尼考胺作为动物体内氟苯尼考的残留标示物[2-3]。 氯霉素对人体的造血系统损害极大,易引起再生障碍性贫血,也可导致致命的“灰婴综合征”[4],我国和欧盟等国家都禁止其在食用的动物源性产品中使用。我国农业农村部250号公告将氯霉素列入了《食品动物中禁止使用的药品及其他化合物清单》[5],不允许在动物食品中有残留。由于甲砜霉素和氟苯尼考抗菌谱与氯霉素相似,较氯霉素其毒性有所降低,抗菌活性和安全性优于氯霉素,目前已广泛应用于兽医临床防治细菌性疾病,但仍然对胚胎有影响[6]。为保障食品安全,我国制定了甲砜霉素和氟苯尼考在动物源性食品中的最大残留限量,GB 31650—2019规定了甲砜霉素和氟苯尼考可用于猪、牛、羊、家禽和鱼,但特别规定“家禽(产蛋禁用)”,最高残留限量分别为50 μg·kg-1和100~3 000 μg·kg-1[7]。
高效液相色谱-串联质谱(High Performance Liquid Chromatography-Tandem Mass Spectrometry,HPLCMS/MS)法为目前检测氯霉素类药物最常用的方法。国内外报道的动物组织中氯霉素类药物的残留检测方法很多[8-11],但存在许多局限性,不能满足当前动物性食品中霉素素类药物残留检测效率的需要。部分方法检测药物残留种类覆盖不全,缺少对氟苯尼考的残留标示物氟苯尼考胺的检测,或针对的动物组织不全,适用范围有限等[12-15]。目前采用HPLC-MS/MS同时对猪肉、鸡肉和鱼肉中氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺残留量分析的检测方法报道较少。本研究在GB/T 22338—2008[16]和GB 31658.5—2021[17]标准方法的基础上进行改进,采用氯霉素-D5作为内标,碱化乙酸乙酯提取,正己烷去除油脂,通过MCX固相萃取柱净化,建立了超高效液相色谱-串联质谱法检测猪肉、鸡肉和鱼肉中氯霉素、甲砜霉素、氟苯尼考和氟苯尼考胺残留的方法。本方法灵敏度高、重现性好、定量准确,适用于大批量猪肉、鸡肉和鱼肉样品中氯霉素类药物的定性、定量分析,为政府加强对违规使用兽药问题的监管提供了技术支持。
1 材料与方法
1.1 材料与仪器
氯霉素-D5(CAS 202480-68-0),BePure公司;氯霉素(CAS 56-75-7,纯度98.0%),First Standard公司;甲砜霉素(CAS 15318-45-3,纯度99.31%)、氟苯尼考(CAS 73231-34-2,纯度99.90%)和氟苯尼考胺(CAS 76639-93-5,纯度99.5%),Dr.Ehrenstorfer公司;乙酸乙酯、乙腈、正己烷,色谱纯,霍尼韦尔贸易(上海)有限公司;乙酸分析纯,天津市康科德科技有限公司;氨水,色谱纯,上海安谱科技股份有限公司;Oasis MCX固相萃取小柱(60 mg/3 mL),美国Waters 公司;猪肉、鸡肉和鱼肉样品采购于某网购平台。
AB Sciex Triple Quad 4500高效液相色谱-串联质谱仪,AB Sciex公司;分析天平,奥豪斯仪器(上海)有限公司;均质机、MS3 digital漩涡混合器,德国IKA公司;Multi Reax涡旋振荡器,德国Heidolph公司;AutoEva-6氮吹仪,杭州奥盛仪器公司;ROTINA 380R 离心机,德国Hettich公司。
1.2 实验方法
1.2.1 试料的制备
取适量新鲜或解冻的空白或供试组织,绞碎,并均质。
1.2.2 提取
称取试样2.0 g于50 mL离心管中,准确加入100 μL 100 ng·mL-1氯霉素-D5溶液,涡旋混匀,静置15 min, 加入乙酸乙酯-氨水(98+2,V/V)溶液10 mL,振荡1 min,10 000 r·min-1离心3 min,转移上清液于另外一只离心管中,残渣重复提取2次,合并3次提取液。离心管中加入5%乙酸2 mL,振荡混匀,于40 ℃ 水浴中浓缩至1.5 mL。转移至50 mL离心管中,用5%乙酸2 mL洗涤氮吹管,洗涤液转移至同一离心管,加正己烷5 mL脱脂,振荡1 min,10 000 r·min-1离心5 min,弃上层,下层提取液重复脱脂一次,备用。
1.2.3 净化
将提取液转移至MCX固相萃取小柱(60 mg/3 mL,使用前依次用2 mL甲醇和2 mL水活化)中,弃去。用2 mL 5%乙酸溶液淋洗,用5 mL 10%氨水甲醇洗脱,收集洗脱液,于40 ℃水浴中氮吹干。用0.5 mL 30%乙腈水溶溶解残渣,混匀后经0.22 μm有机滤膜过滤后供UHPLC-MS/MS分析。
1.2.4 色谱条件
色谱柱:C18柱(100 mm×2.1 mm,1.7 μm);流速: 0.4 mL·min-1;柱温:40 ℃;进样量2 μL;流动相:A为0.1%氨水溶液,B为乙腈。梯度洗脱程序:0.00~1.30 min, 95%A;1.30~5.00 min,95%A→10%A; 5.00~6.00 min,10%A;6.0~6.01 min,5%A; 6.01~7.00 min,95%A。
1.2.5 质谱条件
电喷雾(ESI)离子源,正负离子切换扫描模式;氯霉素、甲砜霉素、氟苯尼考和氯霉素-D5采用负离子扫描方式,多反应监测模式(MRM),气帘气30 psi,碰撞气9 psi,离子化电压-4 500 V,温度550 ℃,喷雾气50 psi,辅助加热器50 psi;氟苯尼考胺采用正离子扫描方式,多反应监测模式(MRM),气帘气30 psi,碰撞气9 psi,离子化电压5 500 V,温度550 ℃,喷雾气50 psi,辅助加热器50 psi;采集参数见表1。
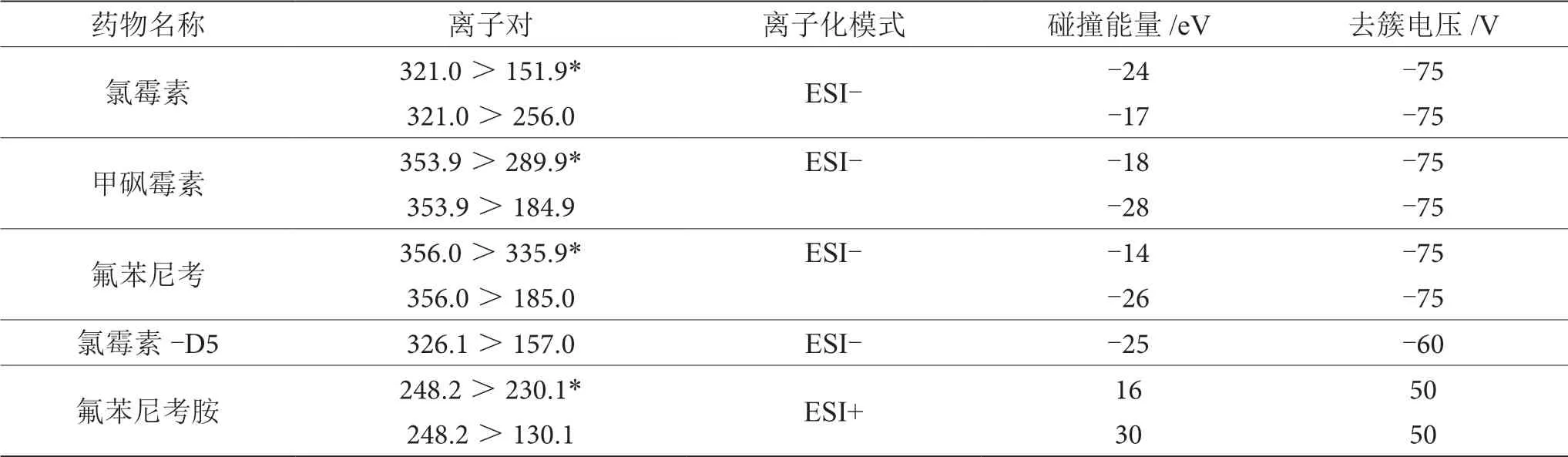
表1 氯霉素类药物信息及质谱参数优化条件表
1.2.6 标准工作曲线的配制
分别称取适量氯霉素类药物标准品于各自的容量瓶中,用乙腈配制成1 mg·L-1的标准储备液。准确移取适量各标准储备液于容量瓶中,用30%乙腈水溶液稀释成1 000 μg·L-1的混合标准中间液。准确移取适量混合标准中间液用相应的空样品基质提取液稀释成浓度为0.5~100.0 μg·L-1的系列混合标准工作溶液,氯霉素-D5内标浓度为20 μg·L-1。
1.3 结果处理
实验结果采用MultiQuant软件进行绘图和数据处理。
2 结果与分析
2.1 液相色谱条件的选择
氯霉素、氟苯尼考和甲砜霉素为弱极性物质,在C18色谱柱上有较强的保留,色谱条件相对容易选择;而氟苯尼考胺为碱性化合物,极性较强在C18柱上保留较弱。本方法比较了水-乙腈、0.1%氨水-乙腈和5 mmol·L-1乙酸铵溶液-乙腈作为流动相时各化合物的分离情况。选择水-乙腈作流动相时,氟苯尼考胺的出峰时间较早,易受杂质干扰,从而影响定量结果。而5 mmol·L-1乙酸铵溶液-乙腈作流动相时,氟苯尼考胺有拖尾现象,峰型不对称。选择0.1%氨水-乙腈作流动相时,调整流动相的pH值,氟苯尼考胺的保留时间为3.48 min,分离度和峰形效果较好。通过对比不同的流动相、色谱柱、初始比例以及流速,最终发现Waters BEH C18(100 mm×2.1 m,1.7 m)对氟苯尼考胺有一定的保留。为更好地分离待测物和样品中的杂质,本方法最终选用乙腈的初始比例为5%,0.1%氨水溶液和乙腈进行梯度分析,样品总的运行时间为7 min,让杂质能够完全流出,避免下一针样品产生柱上污染,降低了氟苯尼考胺的基质效应,本方法在相对较短时间内能较好地分离4种物质,满足对目标物的定性和定量要求。
2.2 质谱条件的选择
UHPLC-MS/MS采用的是电喷雾离子化(ESI)使化合物带电,氯霉素、甲砜霉素和氟苯尼考为酸性化合物,采用负模式电喷雾离子源;氟苯尼考胺含有氨基官能团,选择用正模式电喷雾离子源,进行离子扫描。表1中为确认的质谱采集参数;图1为氯霉素、甲砜霉素、氟苯尼考、氟苯尼考胺和氯霉素-D5的标准品多反应监测模式离子描谱图。
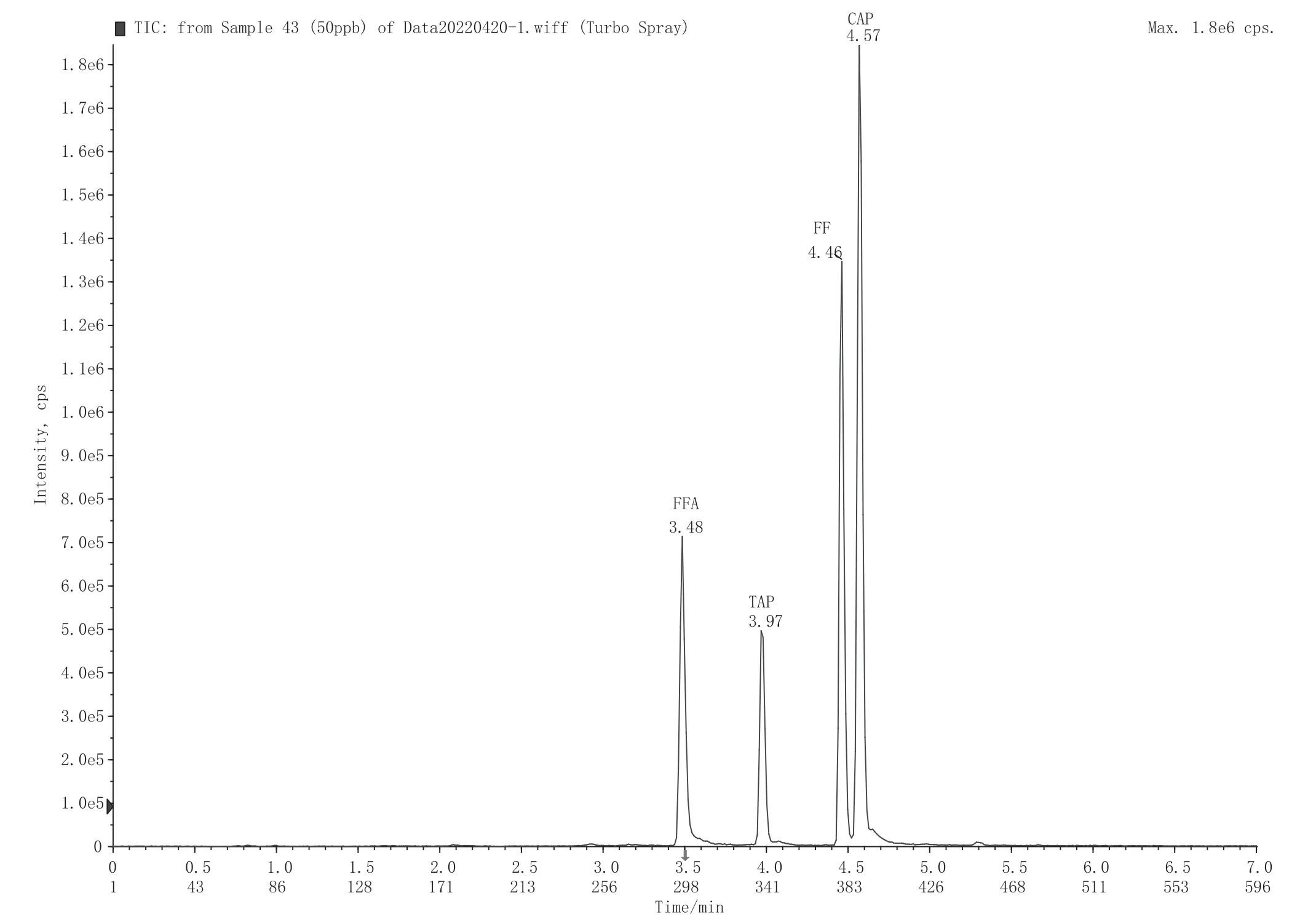
图1 氯霉素药物标准溶液总离子流图
2.3 线性关系、检出限和定量限
分别用3种空白基质提取液配制标准工作曲线,以各待测物与氯霉素-D5的浓度比X为横坐标,以各待测物与氯霉素-D5峰面积比Y为纵坐标,进行回归分析,得到各待测物的回归曲线方程及相关系数 (表2)。由表2看出,4种化合物线性关系良好,相关系数(R2)均大于0.999,可用于准确定量。结合本方法的前处理过程,以3倍信噪比时的响应值计算检出限,本方法氯霉素、甲砜霉素和氟苯尼考的检出限均为 0.1 µg·kg-1,氟苯尼考胺的检出限为0.5 µg·kg-1;以 10倍信噪比时的响应值计算本方法的定量限,4种待测物的定量限均为1.0 µg·kg-1。
2.4 基质效应
基质效应(Matrix Effect,ME)的产生是由于待测物与基质成分中的磷脂、胆固醇等内源性物质一同流出色谱柱,从而产生竞争抑制引起的[18]。在HPLC-MS/MS测定复杂样品时,由于内源性的物质极性较大,会同待测物离子竞相竞争液滴表面,导致待测物的离子化效率降低或增强,引起响应的降低或增高,影响定量的精密度和准确度。分别用3种空白基质提取液配制的标准工作曲线与用30%乙腈水溶液配制的标准工作曲线进行对比,以两条曲线的斜率比值作为考察基质效应强弱的指标,公式为ME=(基质匹配标准工作曲线的斜率/无基质标准工作曲线的斜率-1)×100%。|ME|<20%,为弱基质效应;|ME|为20%~50%,为中等基质效应,而|ME|>50%,为强基质效应。氯霉素类药物的基质效应见表2。从实验结果可以看出,猪肉、鸡肉和鱼肉中氟苯尼考胺的|ME|在20%~50%,为中等基质抑制效应,而氯霉素、甲砜霉素和氟苯尼考的|ME|均小于20%,为弱基质效应。

表2 氯霉素类药物的线性方程、线性范围、相关系数、检出限、定量限与基质效应表
2.5 回收率及精密度实验
分别在猪肉、鸡肉和鱼肉3种不同基质的阴性样品中添加低、中、高3个浓度水平的氯霉素类药物,每个加标水平平行测定6次,计算其回收率和相对标准偏差(Relative Standard Deviation,RSD)。表3的分析结果表明,本方法精密度高(RSD<10%),准确度好,回收率在81%~112%。

表3 氯霉素类药物添加水平、回收率及RSD表(n=6)
3 结论
本方法采用碱化乙酸乙酯提取,正己烷去除油脂,通过MCX固相萃取柱净化,减少基质干扰;采用空白基质提取液配制标准工作曲线以及同位素内标法定量来降低基质效应,以满足测定灵敏度和专属性的要求;通过质谱电喷雾正负离子切换多反应监测模式实现了一针进样4种氯霉素类药物的同时测定。氯霉素、甲砜霉素和氟苯尼考在0.5~100.0 μg·L-1具有良好的线性关系,相关系数(R2)均大于0.999,检出限均为 0.1 μg·kg-1,方法回收率为83%~112%,相对标准偏差小于10%;氟苯尼考胺在1~100 μg·L-1浓度范围内具有良好的线性关系,相关系数(R2)大于0.999,检出限为0.5 μg·kg-1,方法回收率为81%~110%,相对标准偏差小于10%。本方法灵敏度高、重现性好、定量准确,方法检出限能够满足食品安全风险监测的要求,适用于日常大批量猪肉、鸡肉和鱼肉样品中氯霉素类药物及其代谢物残留的定性、定量分析。
猜你喜欢
煤化工(2022年3期)2022-07-08
波谱学杂志(2022年2期)2022-06-14
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
四川农业科技(2021年9期)2021-11-19
食品安全导刊(2021年20期)2021-08-30
染整技术(2019年6期)2019-10-18
分析化学(2019年3期)2019-03-30
山东工业技术(2016年10期)2016-09-06
家庭科学·新健康(2016年5期)2016-05-12
绿色科技(2015年2期)2015-04-22
