
A new global potential energy surface of the ground state of SiH+2(X2A1)system and dynamics calculations of the Si++H2(v0=2,j0=0)→SiH++H reaction
2022-11-21 09:28YongZhang张勇XiugangGuo郭秀刚andHaigangYang杨海刚
Chinese Physics B 2022年11期
关键词:张勇
Yong Zhang(张勇) Xiugang Guo(郭秀刚) and Haigang Yang(杨海刚)
1Department of Physics,Tonghua Normal University,Tonghua 134002,China
2Weifang University of Science and Technology,Shouguang 262700,China
A global potential energy surface(PES)of the ground state of SiH+2 system is built by using neural network method based on 18223 ab initio points. The topographic properties of PES are presented and compared with previous theoretical and experimental studies. The results indicate that the spectroscopic parameters obtained from the new PES are in good agreement with the experimental data. In order to further verify the validity of the new PES,a test dynamics calculation of the Si++H2(v0=2,j0=0)→H+SiH+reaction has been carried out by using the time-dependent wave packet method.The integral cross sections and rate constants are computed for the title reaction. The reasonable dynamical behavior indicates that the newly constructed PES is suitable for relevant dynamics investigations.
Keywords: potential energy surface,integral cross section,rate constant,time-dependent wave packet
1. Introduction
The Si++ H2reaction as one of ion–molecule reactions was received relatively less attention when compared with other reactions, such as O++ H2and C++ H2.[1–4]However, the studies of the Si++ H2reaction and its reverse reaction are of great practical significance, because the SiH+ion is postulated as an intermediate specie in the manufacturing process of amorphous silicon films in chemical vapor deposition process.[5–7]Furthermore, the Si++H2reaction and its reverse reaction are of considerable interest in astrophysics and spectroscopy. For example, the SiH radical and SiH+ion have been observed in the Sun’s photosphere.[8,9]Therefore,the spectroscopic and dynamics information of SiH2and SiH+2systems are very important for the modeling of star atmospheres.
Since Curtis and co-workers[10]reported the ˜A2B1(Π)←˜X2A1transition electronic spectrum of SiH+2ion by using laser photofragment spectroscopy in 1985, numbers of theoretical studies have been carried out. The ˜X2A1and ˜A2B1potential energy surfaces (PESs) of SiH+2ion in collinear geometries have been reported by Hirstet al.[11]in 1986. Later,Gonz´alezet al.[12]also presented a new PES inC∞vandC2vsymmetry and the Si++H2→SiH++H reaction was studied. The PESs of four lowest electronic states of the SiH+2system was constructed by Mort and co-workers[13,14]in 1994. The conical intersection between ground state and excited state has been found and the influence of conical intersection on the photodissociation dynamics has been discussed. In the same year,the three-dimensional potential energy functions of ˜X2A1and ˜A2B1states of the SiH+2system were reported by Baueret al.[15]and rovibronic spectrum of the SiH+2ion was calculated and compared with experimental values.In 2019,a new global PES for the ground state of the SiH+2system was constructed by Gaoet al.[16]with the many-body expansion method. In addition, the H + SiH+→Si++ H2reaction has been studied by using Chebyshev quantum wave packet method based on the new PES.The reaction probabilities,integral cross sections(ICSs)and the rate constants were reported in their work.Later, Zhaoet al.[17]recalculated the H+SiH+→Si++H2reaction based on Gao’s PES.[16]In their work,the Chebyshev quantum wave packet method is used and the full Coriolis coupling effect is considered.The reaction probabilities,ICSs and rate constants were reported and the results indicated that the Coriolis coupling has large effect on the final dynamical results.
As discussed above, much less effort has been made in the construction of global PES and related dynamics calculation. The aim of present work is to construct a new global PES for the ground state of the SiH+2system.In order to verify the validity of the new PES,a test dynamics calculation of the Si++H2(v0=2,j0=0)→H+SiH+reaction will be carried out based on the new PES.In this paper,the methods used in theab initiocalculation,fitting processes and dynamics calculation are shown in Section 2. The topographical features of the PES are presented in Section 3 as well as the dynamical results. The conclusions are displayed in Section 4.
2. Potential energy surface
2.1. Ab initio calculations
Theab initiocalculations are carried out with MOLPRO software package[18]and theCssymmetry is used for all configurations.In theab initiocalculation,the aug-cc-pVQZ basis set[19,20]for H atom and cc-pwCVQZ[21]basis set for Si atom and an internally contracted multi-reference configuration interaction (MRCI)[22]method is used. In detail, we first perform the state-averaged complete active space self-consistent field (SA-CASSCF)[23,24]calculations over three electronic states (two2A′and one2A′′) with equal weight. In addition,15 valence electrons of the SiH+2system were involved 13 active orbitals(11a′+2a′′). In detail,the 1s,2s,and 2p orbitals of Si+are closed which related to the 4a′and 1a′′. The active orbitals 7a′and 1a′′were mainly contributed by 1s orbital of H and 3s,3p orbitals of Si+,while the 2s orbital of H and 4s orbital of Si+occupied a relatively small weight. Then, the MRCI calculations were performed with the reference orbitals which provided by the SA-CASSCF calculation. Finally, a total of 21530ab initiopoints was calculated. In fitting process,theab initiopoints about 10 eV larger than the minimum potential energy were abandoned and 18223ab initiopoints were used to construct the PES. The Jacobi coordinates (RQ,RHH,α) were used to sample theab initiopoints.RQis the distance between Si+ion and the midpoint of H2molecule.RHHis the bond length of H2molecule andαis the angle betweenRQandRHH. In detail, 1.0 Bohr≤RQ≤25.0 Bohr,0.6 Bohr≤RHH≤25.0 Bohr,and 1°≤α ≤89°.
2.2. PES fitting
The permutation invariant polynomial neural network(PIP-NN)[25,26]method is adopted in this work, which is widely used in the PES construction such as BrH2,[27]KH2,[28]and LiH2[29]system. To avoid the discontinuities of the derivatives around the boundary, PIP method is used to transform the three bond lengths of the SiH+2system. In detail,G1=(PSiH+a+PSiH+b)/2,G2=PSiH+a·PSiH+b,G3=PHH,wherePSiH+a= exp(-0.2·RSiH+a),PSiH+b= exp(-0.2·RSiH+b),andPHH=exp(-0.2·RHH),respectively.In the fitting process,G1,G2,andG3are used as the input term and two hidden layers are used, each of which involves 15 neurons. The neural network expansion can be written as


Over-fitting has always been a problem of neural network.To deal with this problem,the input data are randomly divided into three parts,namely,training part(90%),testing part(5%)and validation part(5%).The root means square error(RMSE)is used to test the accuracy of PES and it can be presented as

2.3. Topographical features of the potential energy surface
To test the accuracy of PES, the spectroscopic parameters of SiH+(X1Σ+) ion and H2(X1Σ+g) molecule obtained from the PES are compared with available theoretical[16]and experimental[30,31]values and the results are shown in Table 1.

Table 1. The spectroscopic constants of the SiH+ (X1Σ+)and H2 (X1Σ+g)molecules.
As shown in Table 1,the spectroscopic constants obtained from the PES are in good agreement with experimental data.For the SiH+(X1Σ+) ion, the bond length, harmonic vibrational frequency, and dissociation energy obtained from the PES deviate from the experimental values by 0.0017 Bohr,14.57 cm-1, and 0.015 eV, respectively. It is very clear that present values closer to the experimental data than the results reported by Gaoet al.[16]For the H2molecule,the bond length is same with experimental data, the harmonic vibrational frequency lower than the experimental data by about 2 cm-1,and the dissociation energy is lower than the experimental data by 0.02 eV.As a whole,present PES can give a good description for the two-body term.
The major topographical features of the newly constructed PES are shown in Figs. 2(a) and 2(b), which correspond to the global minimum and vertically approaching geometries,respectively.It is very clear that present PES shows a smooth and correct behavior over the configuration space studied. Figure 2(a)shows the contour plot for the bending angle fixed at 119.73°. Clearly, only a deep potential well is observed and no potential barrier is found. In addition,Fig.2(a)shows a symmetrical behavior which can be attributed to the exchange symmetry of H atom. Figure 2(b)exhibits the contour plot for the Si+ion approach the H2molecule in theC2vsymmetry. Two potential wells are found which correspond to a local minimum and global minimum geometries, respectively. In addition,a potential barrier is also found.
To make a comparison with available theoretical and experimental results,the spectroscopic parameters of theses stationary points are presented in Table 2 as well as the values obtained from previous theoretical and experimental studies.As shown in Table 2,the global minimum is located atRHH=4.8400 Bohr,RSiH+a= 2.8008 Bohr,RSiH+b= 2.8008 Bohr,andθ[HSiH]+=119.73°,which is in good agreement with experimental dataRHH= 4.8529 Bohr,RSiH+a= 2.8161 Bohr,RSiH+b=2.8161 Bohr, andθ[HSiH]+=119°. The recently reported PES by Gaoet al.[16]obtained similar results with ours,both of which are in good agreement with experimental data.For the harmonic vibrational frequency, theωsymandωasymare in good agreement with each other, except for theωbend.For the local minimum,present values are in good agreement with the values reported by Gaoet al.[16]for both geometric structures and harmonic vibrational frequencies. However,in the transition state,great discrepancies can be found between present values and the results obtained from Gaoet al.[16]
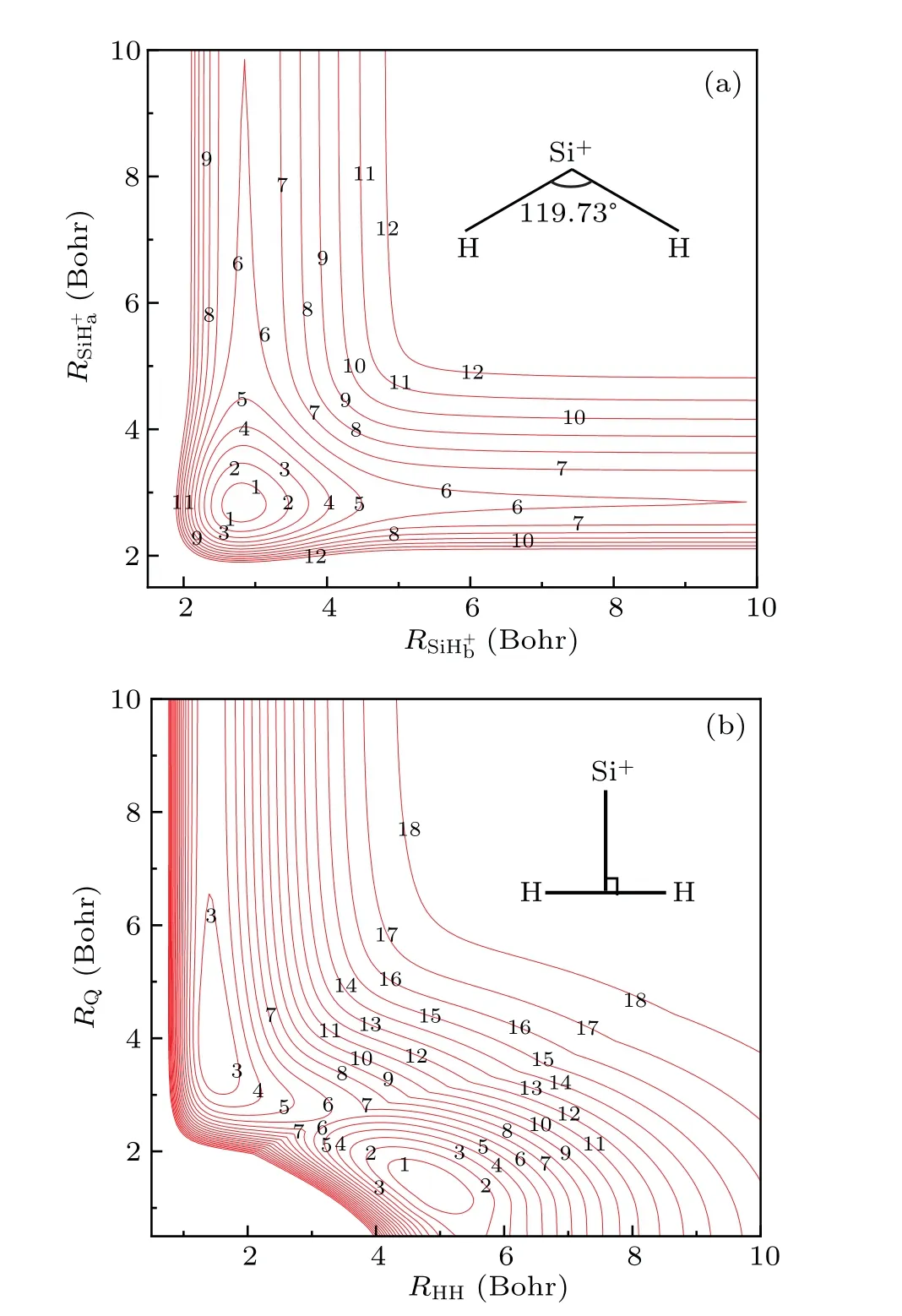
Fig. 1. Equipotential contour plot (a) for the structure of global minimum value in which the angle ∠[HSiH]+ = 119.73°, contours are equally spaced by 0.4 eV, starting at -5.1 eV and (b) for the Si+ ion approach to H2 molecule in C2v symmetry,contours are equally spaced by 0.4 eV,starting at-5.1 eV.
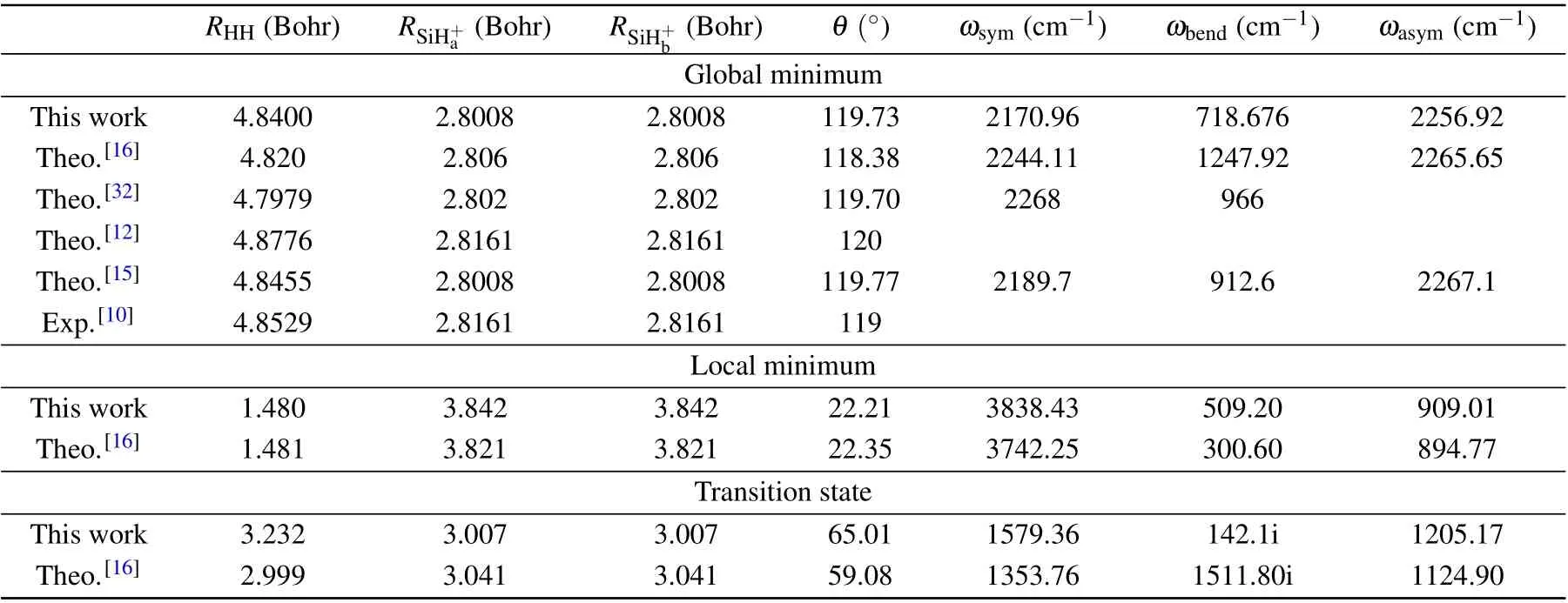
Table 2. The spectroscopic parameters of stationary points.
In order to find the reason why present PES and Gao’s PES are so different in the transition state, Fig. 2 shows the potential energy curves of Si+ions close to the midpoint of H2underC2vsymmetry when the bond length of H2is fixed at 2.999 Bohr and 3.232 Bohr. To compare conveniently, theab initiovalues and the results obtained from Gao’s PES are also displayed in Fig.2. Clearly,there is a conical intersection between the ground state (12A′) and excited state (22A′) and present PES gives a correct description of the conical intersection. However, the potential energy curves obtained from Gao’s PES obviously cannot correctly describe the conical intersection. We suppose this may be attributed to the drawback of many-body expansion method.
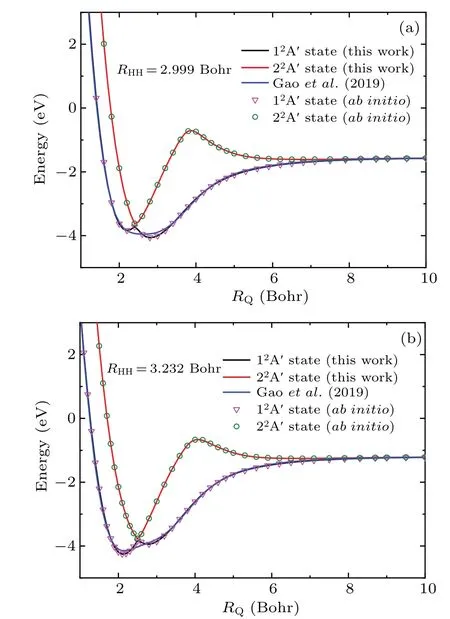
Fig.2. The potential energy curves for the Si+ion approach to the midpoint of H2 molecule at C2v symmetry along with the ab initio results and the values obtained from Gao’s PES[16] ((a)for RHH=2.999 Bohr,(b)for RHH=3.232 Bohr).
Figure 3(a)shows the change of potential energy for the Si+ion moves around the H2molecule as the bond length of H2fixed at 1.401 Bohr. As seen in the figure, there is a potential well with depth of about 0.4 eV at the positionRQ=4 Bohr, andθ=90°. Figure 3(b) exhibits the H atom moving around SiH+ion asRSiH+=2.844 Bohr. Obviously,there is a shallow potential well and a deep potential well on the side of H atom and Si+ion,respectively. The deep well is located atRQ=3.8 Bohr,θ=40°with the depth about 2.7 eV and the shallow well is located atRQ=1.2 Bohr,θ=135°with the depth about 0.9 eV.
Figure 4 displays the minimum energy paths of the PES at several selected angles. As seen in the figure, the energy of Si++ H2asymptote is set to be zero. It is very clear that the Si++H2→SiH++H reaction is highly endothermic and the endoergic energy is about 1.30 eV when zero-point energy is not considered. As seen in the figure, there is a shallow potential well and a deep potential well on both sides of the barrier at 30°. The potential well on the right side gradually disappears with the increases of angle. The barrier decreases at first,then disappears,and then gradually increases with the increasing of angle. In the present work, the spin–orbit coupling effect is not considered since it has a little influence on the endothermic energy of the Si++H2reaction.
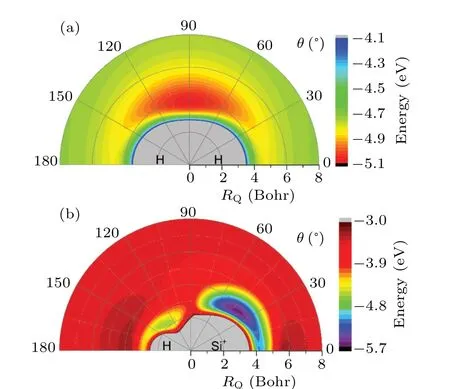
Fig. 3. (a) Color plot for the Si+ ion moves around the H2 molecule when the bond length of H2 is fixed at 1.401 Bohr. (b)Color plot for H atom moves around SiH+ ion when the SiH+ bond length fixed at the equilibrium geometry.
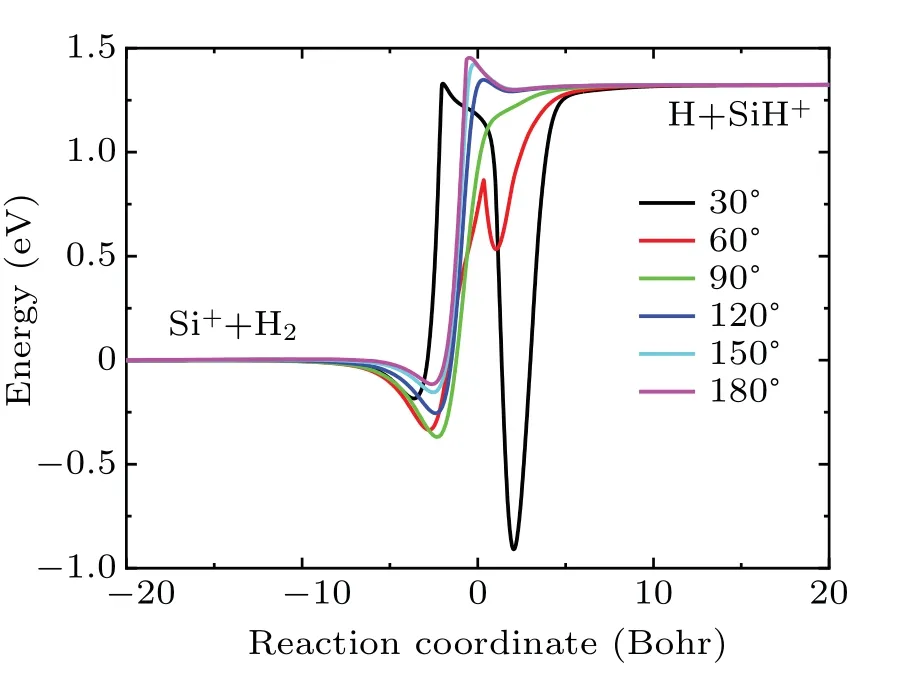
Fig.4.The minimum energy paths of the new PES at different[SiHH]+angles.
3. Dynamics
Based on the newly constructed PES, the dynamics calculations of the Si++ H2→SiH++ H reaction was carried out by using time-dependent wave packet method, which is widely used in the reaction scattering calculation, such as K+ H2,[28]and Li + H2.[29]As discussed above, the Si++H2→SiH++ H reaction is highly endothermic with the energy about 1.3 eV.We suppose the reaction probability of initial state (v0=0,j0=0) will be very small, which is similar with K+H2reaction.[28]Therefore,we chose(v0=2,j0=0)as the initial state. In the dynamical calculations, the convergence test should be carried out at first. In the present work,the convergence test is performed on the reaction probability ofJ=0. Finally, the optimal parameters are obtained and shown in Table 1. As shown in Fig. 4, there is a deep well on the reaction path,therefore it is very time consuming. For reducing the computational cost,the reaction probabilities for particularJ(e.g., 0, 10, 20, ..., 60) were computed and the others were obtained by using theJ-shifting method.[33]

Table 3.Numerical parameters used in the TDWP calculations(atomic units unless otherwise stated).
Figure 5 shows the ICSs of the title reaction in the collision energy range from 0 to 1.0 eV. As seen in the figure,a threshold about 0.19 eV is found which is consistent with Fig.4 as the zero-point energy and vibrational energy considered. In addition,some resonance structures are found on the ICS curve,and this can be attributed to the deep well on the reaction path which support numbers of quasi-bound and bound states. The ICSs increase with the increasing collision energy,which is similar with other endothermic reactions.
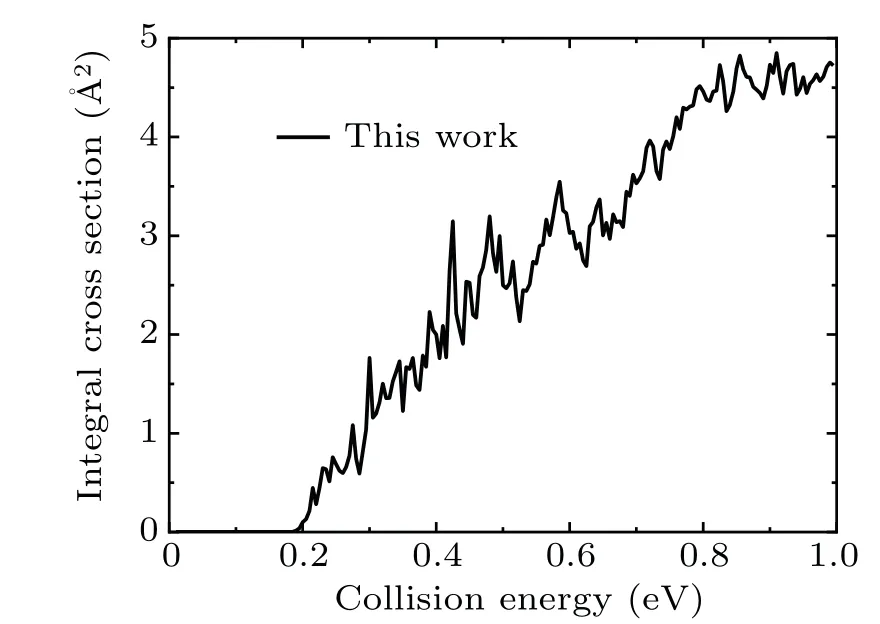
Fig.5. The integral cross sections of the Si++H2 (v0 =2,j0 =0)→SiH++H reaction as a function of collision energy.
The rate constants of the initial state(v0=2,j0=0)are presented in Fig. 6 in the temperature range from 333 K to 1000 K. As shown in the figure, the rate constant increases with roughly a factor about 20 with the increase of temperature. We suppose the rate constants of SiH+2system will be helpful for the modeling of star atmospheres.

Fig.6. The rate constants of the Si++H2 (v0=2,j0=0)→SiH++H reaction in the temperature range up to 1000 K.
4. Conclusions
Based on 18223ab initiopoints, the global PES of the SiH+2system was constructed by using PIP-NN method. The properties of the new PES are discussed and compared in detail with previous theoretical and experimental data. The spectroscopic constants obtained from the new PES are in good agreement with experimental and previous theoretical results.In addition, present PES is in general better than previous ones, and especially the new PES gives a correct description in the region around the conical intersection. To verify the validity of the new PES,the dynamics calculations of the Si++H2(v0=2,j0=0)→SiH++ H reaction is studied by using the TDWP method. The dynamics properties such as ICSs and rate constants are reported in this work. In conclusion,the new PES of the SiH+2system shows reasonable and accurate behavior over the configuration space studied.
Acknowledgement
Project was supported by Key Projects of Science and Technology in the 13th Five Year Plan of Jilin Provincial Department of Education,China(Grant No.JJKH20200482KJ).
猜你喜欢
做人与处世(2022年6期)2022-05-26
做人与处世(2022年4期)2022-05-26
书香两岸(2020年3期)2020-06-29
小猕猴学习画刊·下半月(2019年8期)2019-09-16
理科考试研究·初中(2017年4期)2017-11-04
新高考·高二数学(2014年12期)2015-10-16

- Chinese Physics B的其它文章
- A design of resonant cavity with an improved coupling-adjusting mechanism for the W-band EPR spectrometer
- Photoreflectance system based on vacuum ultraviolet laser at 177.3 nm
- Topological photonic states in gyromagnetic photonic crystals:Physics,properties,and applications
- Structure of continuous matrix product operator for transverse field Ising model: An analytic and numerical study
- Riemann–Hilbert approach and N double-pole solutions for a nonlinear Schr¨odinger-type equation
- Diffusion dynamics in branched spherical structure