
基于Contig的单面基因组片段填充问题研究
2022-11-25 02:56朱永琦李胜华崔晓宇
计算机技术与发展 2022年11期
柳 楠,朱永琦,李胜华,崔晓宇
(山东建筑大学 计算机科学与技术学院,山东 济南 250101)
0 引 言
随着二十世纪三大科学计划之一的人类基因组计划的实施,大量的生物学数据有待处理[1-4],如何利用计算机建模、仿真等技术去提取其中有用的数据,进而研究其中所蕴含的生物学意义,对计算机科学技术来说是一项严峻的挑战[5-6]。因此在二十世纪提出了一门新兴交叉学科—计算生物学。计算生物学运用数学、计算机和生物学相关理论解决生物学问题,已经成为目前最活跃的研究领域之一[7-9]。
基因组片段填充问题[10-12]是计算生物学极其经典的问题之一,其中含重复基因的基因组片段填充问题已经被证明为NP-完全问题[13-14],如何优化基因组片段填充近似算法是近些年来的讨论热点。依据基因样本序列中是否含有重复基因,将该基因组填充问题分为含重复基因的基因组片段填充问题和无重复基因的基因组片段填充问题;或依据基因样本序列不完整数量,将该基因组填充问题分为单面基因组片段填充和双面基因组片段填充,其中一条序列完整,另一条序列缺失,称为单面基因组序列,两条基因序列均为不完整的,则为双面基因组序列[15]。
该文重点讨论单面重复基因组片段填充问题。Munoz和D. Sankoff等人[12-13]首次提出了基于最小重组距离(DCJ距离)的单面基因组填充方法,使用断点图设计了多项式时间算法,并证明了基于DCJ距离的单面基因组片段填充算法是多项式可解的。对于单面无重复基因组片段填充问题,H. Jiang等人提出了使用DCJ距离或断点距离为度量的算法,并证明了其是多项式可解的[14];对于含重复基因的基因组片段填充问题,H. Jiang等人证明了其是NP-完全的,并提出了4/3-近似算法[14-16]。随后N. Liu等人采用局部优化和贪婪算法将该类问题近似度改善到1.25[17-18];J. Ma等人采用非盲局部搜索策略将该类问题近似度进一步改善到1.2[19-20]。
在许多应用中,基因组序列通常被定义为一系列连续的片段重叠群(contig)[21],其中任何一个contig都不能被破坏,缺失基因的插入只能在contig的两端执行。在此约束下,当不存在重复基因时,单面基因组片段填充问题是多项式可解的;当存在重复基因时,H. Jiang等人通过最大化公共邻接证明了该类问题是NP-完全的,并提出了一个近似值为2的近似算法[22-23]和一个双参数的FPT算法[22](k,公共邻接数,d,基因最大重复数);L. Bulteau等人给出了一种基于最大邻接数和最小断点距离的k-Mer参数的FPT算法[24];Q. Feng等人通过构造辅助图和二次寻找最大匹配给出了2.57-近似算法[25]。
该文的主要工作有以下三个方面:系统归纳了基于contig的单面基因组片段填充问题的现有算法并通过实例实现了算法,有助于读者对此类问题的进一步了解;在技术应用和时间复杂度等方面对现有算法做了对比,并分析了该些算法存在的一些弊端;分析接下来研究工作中面对的挑战和可能的解决方案。
1 相关定义
该文只关注基于contig的单面基因组片段填充算法,但其结果可以推广到多染色体或环状基因组。
首先,给出一些必要的定义。不失一般性,假设所有的基因和基因组都由无符号的字母和整数组成,给定一个集合Σ和一个基因序列S,使用c(S)表示基因序列S中所有符号的集合。如果Σ中的符号在基因序列S中出现且只出现一次,则称S是Σ上的一个排列,否则称为序列。对于Σ中的任意两个符号x,y,如果基因序列S至少包含{xy,yx}中的任意一个子集,那么则称x,y在S中邻接,令P(S)为S中所有邻接的集合。设A和B是Σ中的两个基因序列,A={a1a2…an},B={b1b2…bm}。对于P(A)中的任意一个邻接aiai+1和P(B)中的任意一个邻接bjbj+1,如果aiai+1=bjbj+1(或aiai+1=bj+1bj),则称aiai+1与bjbj+1构成了公共邻接,a(A,B)表示A和B的公共邻接集合,同时称(aiai+1,bjbj+1)为一个匹配对。如果P(A)和P(B)中不存在aiai+1=bjbj+1(或aiai+1=bj+1bj),则称aiai+1相对于bjbj+1构成了断点,bp(A,B)和bp(B,A)分别表示A和B的断点集合,如图1所示。
定义一个基因组序列是由一系列contig构成的,且contig内部不能插入缺失基因,即S=
下面具体给出One-Sided-SF-max问题的概念:
定义1:One-Sided-SF-max问题。
输入:一个完整的基因组序列G和一个不完整的基因组序列S,其中S=
输出:将X=c(G)-c(S)≠Ø插入S得到S',使得|a(S',G)|最大。
2 One-Sided-SF-max问题
One-Sided-SF-max问题已经被证明为NP-完全的[22],此类问题不能在有效时间内求出精确解,因此设计近似算法更具有实际意义。本节主要对One-Sided-SF-max问题进行简要介绍,概括分析了国内外经典的三种算法:2-近似算法、2.57-近似算法以及k-Mer算法。
2.1 One-Sided-SF-max问题的2-近似算法
该算法由H. Jiang等人提出,主要使用了贪婪和最大匹配的思想来实现基于contig的单面基因组片段填充。首先在该算法中给出以下定义:对于基因序列S=
定义缺失基因集合X中有一个长度为n的子串,如果插入到slot <βi,αi+1>中(1≤i≤m-1),产生n+1个新公共邻接,称子串为n-Type-1类型串。同样的,产生n个新公共邻接,称为n-Type-2类型串;产生n-1个新公共邻接,称为n-Type-3类型串。算法大体流程如下:
(1)计算缺失基因集合X=c(G)-c(S);
(2)对于缺失基因集合,采用贪婪策略将1-Type-1类型串插入到相应的slot中,并将该slot锁定,不允许其他缺失基因插入;
(3)构造二分图并求其最大匹配,将1-Type-2类型串插入到可构成外部邻接的slot处,并对该slot进行更新:如果xj插入到slot •ai前面,那么将此slot更新为•xj,如果•xj插入到slotβi•后面,那么将此slot更新为xj•;
(4)以步骤2后的缺失基因为顶点构造多重图:若x∈X,y∈X且xy为G中一个内部邻接,那么x和y之间添加一条边,寻找最大匹配M。对于最大匹配M中的所有匹配对xy,如果x为步骤3中插入的元素,则将y插入到相应slot处使得xy构成邻接;将其余匹配对xy任意插入到未锁定slot中,且不能破坏现有邻接;
(5)在不破坏现有邻接关系的前提下,将所有剩余缺失基因任意插入到S中未锁定的slot处;
(6)得到近似解S'。
对于该算法,下面通过一个实例(如图2所示)来说明算法的执行过程:
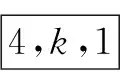
(2)搜寻1-Type-2类型串,找到2,d,g为1-Type-2类型串,并以1-Type-2类型串集合和未锁定slot集合为顶点,构造二分图,建立的二分图BG1如图3所示。
(3)以步骤2之后的剩余缺失基因集合为顶点构造多重图Q,建立的多重图Q并求得最大匹配,将a插入到g之前,将7插入到d之后,将52插入到a之前。
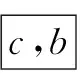
(5)算法结束,得到填充后的基因序列S'=
<52ag4k1acbd21d727>。
以上可以看出,通过此算法可以得到8个新公共邻接,同时此例的最优解为S*=<2ag4k1acb1d7527d2>,有13个新公共邻接。
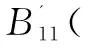
所以,
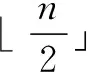
所以有近似解:
该算法为一个2-近似算法,同时该算法的运行时间主要由步骤2中计算O(n)个顶点的二分图中的最大匹配以及步骤3中计算O(n)个顶点的多重图中的最大匹配决定,两者都需要O(n2.5)个时间,所以2-近似算法的时间复杂度为O(n2.5)。
2.2 One-Sided-SF-max问题的2.57-近似算法
Q. Feng提出的2.57-近似算法继续考虑了冗余块对填充过程存在的影响。该算法主要使用了最大匹配算法构造简单路径来具体实现基于contig的单面基因组片段填充问题。
首先在该算法中给出以下定义:令F(S)为contigCi的首尾元素集合,F(S)=(α1,β1,…,αm,βm)。如果最大匹配M中有块xy,xy在最大匹配M中出现的次数称为xy的指示数。设xy与bp(G,S)中块ab可构成匹配对(xy,ab),若xy在M中出现次数大于ab在bp(G,S)中出现次数,则称块xy为一个冗余块。定义⊕为对称差,A⊕B=(AB)∪(BA),令K'为此算法中的两次最大匹配M1和M2的对称差,则K'中每个连通分量必为简单路径或简单循环。算法的大体流程如下:
(1)计算缺失基因集合X=c(G)-c(S)和断点集合bp(G,S);
(2)基于缺失基因集合X、断点集合bp(G,S)和S中每个contig首尾元素集合F(S)构造一般图Γ1,寻找最大匹配M1;
(3)对于M1中的任意块xy,有以下三种情况:如果x和y分别为同一个slot的前后两端,则合并此相邻的两个contig;如果x(或y)属于F(S),将y(或x)插入相应的slot中使得xy邻接;如果x和y均不属于F(S),则将其置于图H'中顶点。依据以上更新基因序列为S1;
(4)基于G、S1和H',求得断点集合bp(G,S1)和F(S1);
(5)更新图H':删除可与bp(G,S1)中断点构成匹配对的边;
(6)基于缺失基因集合X、断点集合bp(G,S1)和集合F(S1)构造一般图Γ2,寻找最大匹配M2;
(7)Δ=H'⊕M2;
(8)对于图Δ中的任意路径k=p1p2…pt-1pt,判断其是否为简单路径:若为简单路径,插入到相应slot中,反之删除路径k中任意一条边得到新的路径p1p2…pt-1pt,将路径p1p2…pt-1pt插入到基因序列的最右侧;更新基因序列为S2;
(9)统一将c(G)-c(S2)插入到序列S2的最右侧;
(10)得到填充完成后的基因序列S'。
下面通过上述2-近似算法的同一个实例(见图2)来说明算法的执行过程:
(1)计算断点集合bp(G,S)=<1a,ac,1d,d7,75,52,24,4g,ga,a2,2k,k7,7d,d2>,计算S中每个contig首尾元素集合F(S)=<4,1,c,b,1,1>;
(2)构造图Γ1:X∪F(S)中的所有元素被视为顶点,对于其中任意两个元素x,y,如果有x∈X,y∈X或y∈F(S)且存在一个断点β使得与xy构成一个匹配对,则在x与y之间添加一条边;如果有x,y∈F(S),假设x在contigC1中且为C1中最后一个元素,y在contigC2中且为C2中第一个元素,C1和C2相邻且存在一个断点β使得与xy构成一个匹配对,则在x与y之间添加一条边。在图Γ1中寻找最大匹配M1,如图4所示;
(3)删除M1中的冗余块:对于M1中的块xy,xy与断点ω可构成匹配对(xy,ω),如果xy在M1中的出现次数大于ω在bp(G,S)中的出现次数,则称块xy为冗余块并将其删除;
(4)令{α1,α2,…,αr}为M1中删除冗余块后剩余块的集合,H'为{α1,α2,…,αr}中的块与bp(G,S)中的断点构成匹配对的集合,则此例中H'={(ac↔ac),(d1↔1d),(52↔52),(g4↔4g),(a2↔a2),(d2↔d2)};
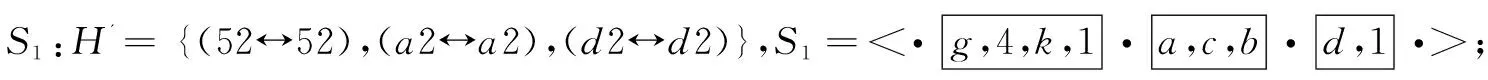
(7)计算X'=c(G)-c(S1)得到缺失基因集合X'=<7,5,2,a,2,7,d,2>,计算S1中每个contig首尾元素集合F(S1)=
(8)计算新的断点集合bp(G,S1)=
(9)使用构造图Γ1的同样方法构造图Γ2;
(10)如果图Γ2与图H'中存在相同边,则在Γ2中删除此条边,此例中,无此类边;
(11)在图Γ2中求得最大匹配M2,如图5所示。
(12)令Δ=H'⊕M2,此例中Δ=(a2,ag,7d);
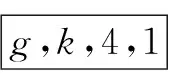

(15)将剩余缺失基因插入到序列S2的最右侧;
(16)得到填充完成后的基因序列S'=
该算法可以得到8个公共邻接,同时其中一个最优解为<2ag4k1acb1d7527d2>,有13个新公共邻接。
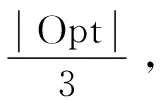
该算法的近似性能比为2.57。
2.3 One-Sided-SF-max问题的k-Mer近似算法
k-Mer算法从参数化复杂性的角度研究基因组填充问题,相较于H. Jiang等人提出的2-近似算法,主要有以下三个方面的不同:
(1)不再限制插入的基因集合为c(G)-c(S),插入集合可以包含比c(G)-c(S)更多或更少的基因集合;
(2)允许将要插入的字符串数量预先指定为输入约束,t1为要插入的字符串数量的下限,t2为要插入的字符串数量的上限(t1≤t2);
(3)作为相似性度量依据,不局限于最大化公共邻接的数量,相反,对于一个预定的参数k,最大化公共k-mers的数目,k值越高,结果越准确。
L.Bulteau等人对于此类问题给出以下定义:对于两个基因序列G和S,G∘S表示二者的串联。存在一个正整数k,使得ak(G)={S[i,i+k]|i∈[n-k]}为序列G中k-mers的集合,则ak(G,S)=ak(G)∩ak(S)。设S[i]表示S中第i个元素,S[i,j]表示序列S中从位置i到j的基因元素。对于一个完整基因序列G和一个不完整基因序列S,令pk(S,G)=ak(G)ak(S)表示存在于G中但不存在于S中的k-mers集合,并将此类k-mers称为潜在的公共k-mers。
定义2:k-Mer Scaffold Filling(k-Mer-SF)。
输入:一个完整的基因组序列G和一个不完整的基因组序列S,其中S=
输出:找到T'⊆T,t1≤|T'|且填充后的S'∈S+T',使得|ak(S',G)|最大。
该文给出一个k-Mer-SF实例,在此只举例说明了k=2和k=3的填充情况(如图6所示)。
H. Jiang等人提出的2-近似算法和Q. Feng等人提出的2.57-近似算法均为k-Mer-SF中t1=t2=|T|且k=2时的特殊情况,并可在多项式时间O(3·(+m)2)内计算。k-Mer-SF相较于以上两个近似算法,不再仅仅参考公共邻接数目的多少,主要使用以下评估参数:k,k-mers的长度;:=ak(S*,G)-ak(S,G),匹配后带来的额外公共k-mers数目;d,一个基因在G中出现的最大次数;m,S中重叠群的个数;t2,要插入的字符串数量的上限;λ,T中字符串长度的上界。
k-Mer算法主要解决了基于动态规划如何在2O()·nO()时间内求得近似解并给出其参数为k+的FPT算法。首先使用着色法对k-mers进行分类,β:T→[t2]表示潜在的公共k-mers,β:T→[t2]表示可能插入字符。图着色后,使用动态规划算法重建序列S,使得S中有个潜在的公共k-mers转为已实现的k-mers,在动态规划过程中逐步找到大小递增的局部最优解,从左到右依次将缺失基因插入到序列S中,并使用局部优化策略避免一些缺失基因重复插入。
对于k-Mer-SF问题,首先对序列G,S及插入基因集合T做约简操作。删除T中多余基因元素,如果T中存在一个基因元素出现次数大于t2,将删除一个元素;其次,对G中的邻接关系分类,并只保留潜在公共邻接,假设x为不出现在S和T中的基因,y为不出现在G中的基因,令P1∘x∘P2∘…∘Pq-1∘x∘Pq替换G且用Ci[1]∘y∘Ci[|Ci|]表示S。如果有一个潜在邻接在G中出现次,则在G中删除一个该邻接,删除后若邻接满足以下条件之一,则称为可实现邻接:
(1)b∈T且c∈T;
(2)存在一个contigCi使得b∈Ci[|Ci|]且c∈T;
(3)存在一个contigCi使得b∈Ci[|Ci|]和c∈Ci[1];
(4)存在一个contigCi使得b∈T且c∈Ci[1]。
建立邻接图H=(V,E):令T,G和S中的基因元素作为H中的顶点,如果邻接bc或邻接cb均为可实现邻接,则令两个顶点b和c相邻,求得最大匹配M。
令V(M)表示匹配的端点,建立两个二分图H1和H2且顶点分别为B:=V(M)和C:=(VV(M))。在H1中,当bc是一个可实现邻接时,在b∈B和c∈C之间添加一条边。在H2中,当cb是一个可实现邻接时,在b∈B和c∈C之间添加一条边。如果H1中存在顶点b∈B且度数至少为2+m+1,则将邻接bc从G中移除;如果H2中存在顶点b∈B且度数至少为2+m+1,则将邻接bc从G中移除,其中c是b的任意邻接。
完成约简操作后,这些顶点的数量最多为|V(M)|·2·(2+m+1)。这给出了G中顶点数量的界限,从而给出了实例大小的界限且所有约简规则可以在多项式时间内执行。
从更广泛的角度来看,k-Mer-SF考虑了字符串的基本插入问题。事实上,可以将该算法扩展到更一般的情况,即给定一个字符串G和一个部分字符串S,完成部分字符串S的插入得到新的字符串S',使得G和S'的相似度最优,因此其包含了H. Jiang等人的问题作为特例。
3 One-Sided-SF-max问题的总结
该文发现2-近似算法没有考虑断点对填充过程的影响和长度大于1的缺失串的插入情况,2.57-近似算法则没有考虑n-Type-3类型串的插入情况,k-Mer算法也仅仅介绍了固定基因子串长度的一般处理情况,然而不同长度以及不同类型串的插入都会对公共邻接数造成影响,从而影响到算法近似比。

序列中存在连续长缺失基因串,在完成1-Type-1类型串的插入后,假设存在可构成n-Type-1类型串的缺失基因串a1a2…an,如若按照2-近似算法中分别处理,则该缺失基因串中1-Type-1类型串被破坏,有以下2种情况:
(1)最多有n个1-Type-2类型串,产生n个邻接;
(2)最少有2个1-Type-2类型串,剩余为n-Type-3类型串,产生2个邻接。
产生邻接数k为2≤k≤n,若将其合并后插入基因序列中,则会产生n+1个邻接。以上证明合并插入n-Type-1类型串具有更优效果。
在处理n-Type-2类型串时,假设存在可构成n-Type-2类型串的缺失基因串b1b2…bn,如若按照2-近似算法分别处理,则该缺失基因串中n-Type-2类型串被破坏,有以下2种情况:
(1)最多有2个1-Type-2类型串,同时bi(2≤i≤n-1)均可与其相邻缺失基因bi-1或bi+1(2≤i≤n-1)构成内部邻接,产生n个邻接;
(2)最少有1个1-Type-2类型串,同时剩余n-1个缺失基因均为n-Type-3类型串,产生1个邻接;产生邻接数k为1≤k≤n,如若将其合并后插入基因序列中,会产生n个邻接。以上证明合并插入n-Type-2类型串具有更优效果。
对于2.57-近似算法,虽然n-Type-3类型串会产生n-1个邻接,但是如果不对其进行处理,任其插入未锁定的slot,存在破坏已有邻接的可能,其中最多会破坏2个邻接,n-Type-3类型串产生邻接数k为n-3≤k≤n-1,明显降低最后的填充效率,所以对其进行插入处理十分必要。
k-Mer算法固定了基因子串的长度,2-近似算法就是一个特例,其固定长度为2的公共邻接数目作为算法性能参考依据,以上也证明了此时并不是最优算法,进而说明了基因填充过程中限制基因子串长度存在影响近似性能比的可能。
4 总结与展望
重点介绍了基于contig的单面重复基因组片段填充问题的研究现状。对该些算法在近似性能比等方面做出了详细的对比(见表1),直观地看出各个算法现存在的一定不足,说明此类算法仍有改进空间,同时提出了该类问题的改进思路。

表1 三种算法分析比较
4.1 面临的挑战
(1)现有算法依赖于公共邻接数目,邻接的定义没有考虑基因序列不存在逆序关系的情况,因此存在两个基因序列的邻接均为公共邻接但二者完全不相似的情况,不利于后续对算法近似比的研究。
(2)现有算法对度量依据的选择较少,并过度依赖于最大匹配算法,因此对此类问题的研究较为片面。
4.2 前景展望
针对目前单面重复基因组片段填充问题的研究工作,发现基因组填充问题有以下发展前景。
(1)目前One-Sided-SF-max问题的最佳性能近似比为2,日后还需要进一步优化。
(2)现有算法均是在最大化邻接基础上考虑其近似比,基于最小断点数层面有待研究。
(3)双面基因组填充问题也被证为NP-完全的,但没有提出近似比算法,此类算法可推广到双面基因组填充问题,有利于双面此问题的近似比优化。
猜你喜欢
学苑创造·C版(2022年8期)2022-06-18
辽河(2022年4期)2022-06-09
中等数学(2021年9期)2021-11-22
中等数学(2021年8期)2021-11-22
艺术评论(2020年5期)2021-01-23
电脑报(2019年20期)2019-09-10
初中生世界·九年级(2019年6期)2019-08-15
好孩子画报(2019年2期)2019-06-11
汽车文摘(2017年4期)2017-12-07
山东工业技术(2016年15期)2016-12-01
