
基于Snakemake的RNA-seq数据自动化分析流程RNApipe
2022-12-03 06:27武乐李益楠孔德信周志鹏
华中农业大学学报(自然科学版) 2022年6期
武乐,李益楠,孔德信,周志鹏
1.华中农业大学信息学院,武汉 430070;2.华中农业大学生命科学技术学院,武汉 430070
转录组测序(RNA-seq)是一项对特定组织或细胞中所有的RNA 进行高通量测序的技术。近十年来,随着测序技术的发展、测序成本的降低以及分析工具的快速开发,RNA-seq 已广泛应用于研究基因表达调控、可变剪切甚至RNA 结构等方向,成为生命科学基础研究领域中非常重要的技术[1]。RNAseq最常应用于识别特定条件下差异表达的基因(dif⁃ferential gene expression,DGE)并 鉴 定 其 分 子 功能[2]。DGE 的分析流程主要包括5 个步骤:对高通量测序平台产生的原始读段(reads)进行质量检测和控制、将预处理后的reads比对到参考基因组、在基因组或转录组水平上进行定量、鉴定样本间差异表达的基因,以及表征差异表达基因的生物学功能[3]。
目前,RNA-seq 的各个分析步骤都已有许多成熟的计算方法。然而,这种分段分析的方法通常会涉及到不同的操作系统和编程语言,这对于科研人员的编程能力提出较高的要求。之前已有多项研究将各个分析步骤集成到自动化分析工作流程中,从而降低对用户编程能力的要求并有效地提高其工作效率。然而,目前已开发的RNA-seq 自动化分析工作流程中存在着一些不足之处。例如,VIPER[4]与hppRNA[5]等工具只支持少数几个特定物种的数据分析,IRIS-EDA[6]与ideal[7]等工具仅适用于差异表达基因的下游分析和可视化,BioJupies[8]与RNA⁃Cocktail[9]等工具不支持原始数据的预处理和质量控制等步骤,而ARRMOR[10]与RASflow[11]等工具缺乏对差异基因的功能注释。
本研究通过Conda 部署环境以确保整个工作流程中的软件快速、平稳地安装,并通过Snakemake 工作流程管理系统集成各个分析步骤,最终构建了1个流程更完整且更易使用的RNA-seq 自动化分析工具——RNApipe,旨在为研究人员快速、简便地从大型RNA-seq 测序数据中获取基本信息提供技术支持。
1 材料与方法
1.1 Conda 环境与Snakemake 工作流程管理系统
对于编程经验有限的用户,安装并维护RNAseq 数据分析中必需的工具及其依赖项通常具有挑战性。Conda作为软件包和环境管理系统,可以自动安装、运行和更新软件包及其依赖项,并且支持虚拟环境的创建、切换与移植,是确保软件管理可持续和可重现的理想工具[12]。因此,RNApipe 使用Conda安装和运行软件,并创建虚拟环境。这不仅能够保证RNApipe 的顺利安装与平稳运行,还确保了独立于操作系统和机器的数据可重复性分析。
为减少数据分析过程中手动执行的步骤,传统的工作管道通常使用自定义的脚本将多个工具进行链接,然而这种管道通常与本地计算基础架构高度相关,难以共享与维护,产生的结果也较难重复[13]。为此,RNApipe 使用Snakemake 工作流程管理系统将RNA-seq 各个分析步骤进行组合。Snakemake[14]是一个基于python 的工作流程管理系统,它具有以下优势:通过集成Conda包管理器自动安装和配置软件,以确保不同平台的可移植性;通过支持高性能并行计算和云计算环境,以实现超越本地基础架构的可扩展性;通过支持工作流间缓存,避免在管道之间重新运行程序而浪费时间和计算资源;并通过将各个步骤分解为模块化规则(rules),从而降低了代码的复杂性,使代码易读易维护易扩展。最终,RNApipe 基于Snakemake 管理系统,可以实现通过1条命令执行整个自动化分析过程,极大地简化了程序的运行方式(图1)。
1.2 RNApipe工作流程框架
整个RNApipe工作流程(图1)包括:(1)RNApipe启动时,首先使用fastp[15](默认)或Cutadapt[16]对每个样本进行检测并修剪接头、去除低质量碱基和reads;随后使用FastQC[17]检测reads 质量,质检结果由MultiQC[18]汇总为一个可视化报告。(2)质量达到用 户 要 求 的reads 会 被HISAT2[19](默 认)或STAR[20]比 对 到 参 考 基 因 组,比 对 结 果 由Quali⁃map2[21]进行质量评估,比对文件以BAM 或Bigwig格式(可选)保存。(3)使用featureCounts[22](默认)或HTSeq-count[23]对BAM 文件进行定量(count)。(4)其中表达矩阵(count)用于DESeq2[24]、edgeR[25](可选)或limma[26](可选)鉴定差异表达的基因。TPM 标准化的表达矩阵用于样本间基因的表达量分析。(5)使用KOBAS-i[27]对差异基因进行GO 功能注释与KEGG pathway 通路分析,对所有表达的基因做GSEA 基因集富集分析。以上每个步骤都可以输出高质量的可视化图片或报告,并保留重要的输出文件和表格数据。
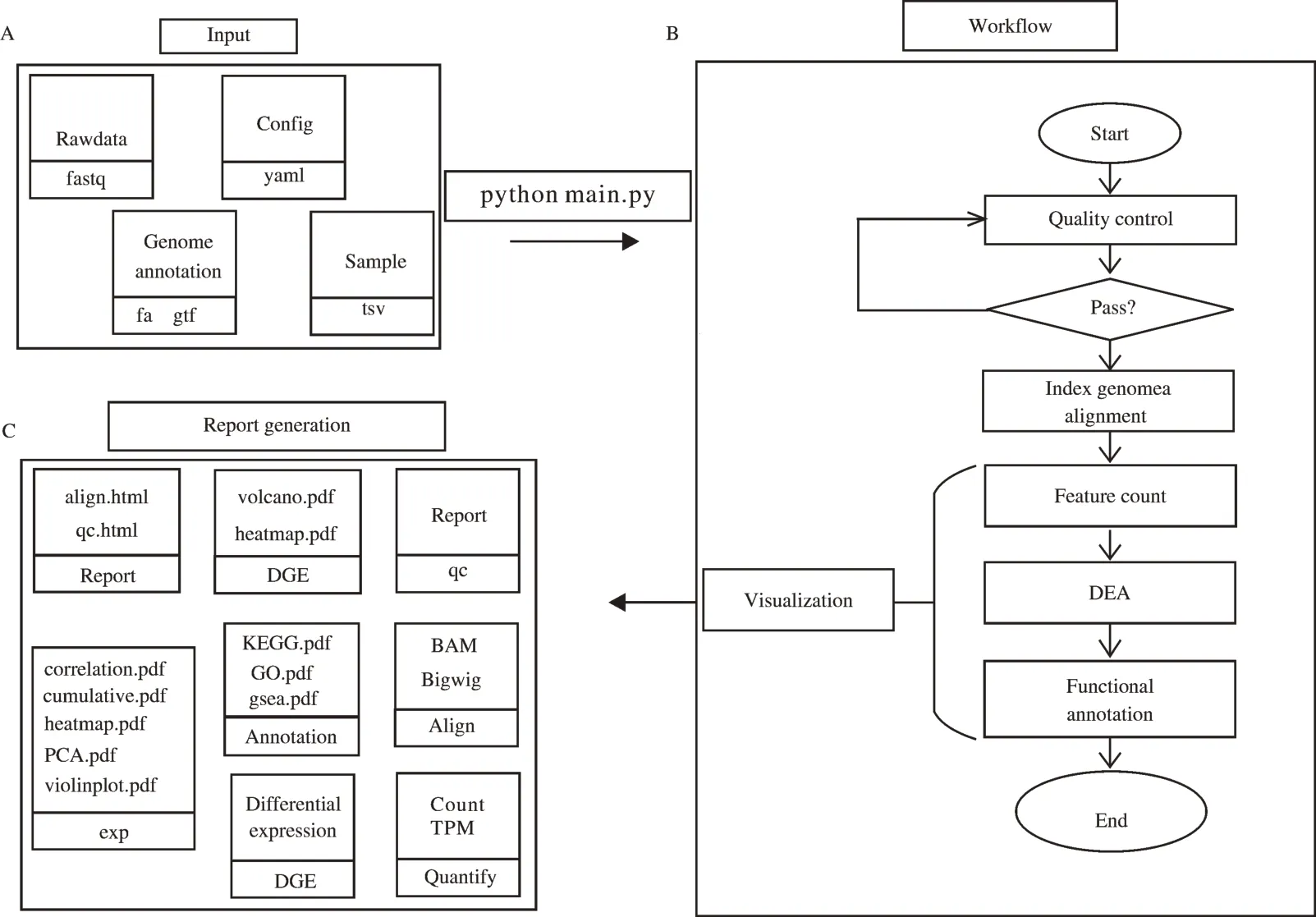
图1 RNApipe数据分析流程图Fig.1 The workflow chart of RNApipe analysis
1.3 RNApipe使用方法
RNApipe 的源代码已经上传至Github 供用户下载 和 使 用:https://github.com/ywu019/RNApipe.git。以下简单介绍RNApipe 的安装和使用方法,具体使用方法可见Github 中的说明文档(Documenta⁃tion.pdf)。
1)下载RNApipe 工具与安装环境。首先通过Python3.7 在Linux 系统中安装miniConda3 环境,随后通过以下3 条命令完成RNApipe 工具的安装与环境 的 创 建:①从 GitHub 下 载 RNApipe:it clonehttps://github.com/ywu019/RNApipe.git;②创建环境:conda env create -n RNApipe -f envs/envs.yaml;③激活环境:conda activate RNApipe。
2)用户指定输入文件。在运行RNApipe 前需要指定:基因组文件(.fa)、注释文件(.gtf)与压缩的测序文件(.fastq.gz)(图1A)。其中基因组文件和注释文件建议从NCBI下载,测序文件支持单端和双端测序,其命名应符合“条件_重复”(或“条件_重复_双端”)的形式,例如root_1_R1.fastq.gz。
随后修改configs/目录下的样本表(samples.tsv)和配置文件(config.yaml)。用户可以根据实验条件指定样本表中的条件列“condition”与重复列“repli⁃cate”的信息,并修改配置文件中的基本设置,配置文件主要包含以下变量:
PROJECT:#项目名称;
READPATH:#测序文件fastq.gz的路径;
END:#测序数据类型是SE(single-end)还是PE(paired-end);
OUTPUTPATH:#输出文件的路径;
GENOME:#基因组文件路径;
ANNOTATION:#注释文件路径;
CONTROL:#对照组名称,与samples.tsv 中condition一致;
TREAT:#处理组名称;
SPEICES_ABBREVIATION:物种简称,在speices_abbreviations中查询;
TRIM:fastp #[fastp,cutadapt]选择工具执行数据预处理;
ADAPTER:AGATCGGAAGAGC # 接 头序列;
ALIGN:hisat2#[hisat2,STAR]#比对软件;
BIGWIG:["no"]#[yes,no]#是否将bam 格式的文件转化为bigwig格式;
DEA:DESeq2 #[DESeq2,edgeR,limma]#差异基因分析软件;
QUANTIFY:featureCounts #[featureCounts,htseq]#定量软件;
THREAD:"10"#线程数目;
TIME:"5"#程序延迟时间。
3)运行工作流程。准备好所有的输入文件后,用户只需要通过一条命令(python main.py)即可在本地或集群环境中运行RNApipe 整个工作流程。运行结果保存在用户指定的输出目录中,logs/目录下生成各个分析步骤的日志文件,其中记录了程序的运行时间、标准输出和标准错误等信息。
2 结果与分析
为了让用户了解RNApipe 的工作原理和基本功能,本研究利用RNApipe 对来自4 日龄野生型拟南芥根部(root)和芽部(shoot)的单端RNA-seq 完整数据集(PRJNA321304;GEO:GSE81332)进行自动化分析,其中根部设为对照组,芽部设为试验组。
2.1 RNApipe概要
图2A 展示了用户从Github 下载的RNApipe 所包含的完整数据集。example_data/目录代表1 个真实的测试数据子集(GSE81332),便于用户在运行实际项目前对系统和软件进行测试。Documentation.pdf 文档对RNApipe 的安装、使用方法以及注意事项进行了详细的介绍。
用户在运行项目时应在project/目录中设置类似于example_data/的项目结构,并将测序数据、参考基因组和注释文件分别置于相应的子目录中(图2A),再根据实验条件修改config.yaml文件(图2B)和sam⁃ple.tsv文件(图2C)。当输入文件设置完毕,只需要1条简单的命令(图1 命令),RNApipe 将根据config.yaml 文件中的设置,通过Snakemake 对main.py 中的规则自动执行整个分析过程。输出结果存于trim/、align/、quantify/、DEA/、annotation/、visualize/等目录,分别对应质控、比对、定量、差异分析、功能注释和图形可视化结果(图1C)。其余文件则不需要修改,除非用户需要自定义工作流程。

图2 RNApipe的文件与目录结构Fig.2 The file and directory structures of RNApipe
2.2 数据质量可视化
为了保证分析结果的准确性,RNApipe 分别对质控后的数据和比对文件进行质量检测,并将每个样本文件生成的质控报告(html)进行汇总(图3)。通过质控报告检查当前reads 是否仍需要修剪,当数据质量较低时,例如:碱基质量差、GC 含量不稳定、存在接头等,则建议继续修剪以提高数据质量。如图3A 所示,6 个样本在质控后的碱基质量均在Q30以上,表明碱基质量较好,可用于后续数据分析。图3B展示了6个样本的详细比对情况。
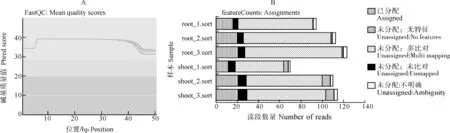
图3 数据质量检测报告Fig.3 Report on data quality inspection
2.3 基因表达量可视化
在完成质量控制和比对后,RNApipe 将表达矩阵(count)进行TPM 标准化,以消除测序深度和基因长度对表达量的影响。设置阈值(TPM<1)过滤低表达的基因,得到的表达量矩阵用于可视化图表展示(图4)。
RNApipe 使用cor 函数与ggplot2 包(v3.3.3)将所有样本基因表达谱的成对相关性可视化为热图,相关系数的绝对值越接近1,表明样本之间的相关性越高,这有助于用户进一步识别相关性较低的离群值样本。结果显示拟南芥根部或芽部中各个生物学重复间的相关性较高,而样本间的相关性较差(图4A)。此外,RNApipe 使用FactoMineR(v2.4)和fac⁃toExtra(v1.0.7)包进行主成分分析(PCA),通过将数据降为二维,再进行K-means 聚类和计算欧式距离,距离值越小则表示数据的相似度越高,用户可以据此分离和排除离群值。PCA 结果同样显示组内的生物学重复聚类良好,而样本间差异较大(图4B)。RNApipe还支持使用pheatmap包(v1.0.12)展示样本中所有表达基因的聚类图谱,横轴顶部的线图表示样本的聚类,左侧线图则表示基因的聚类。此外,RNApipe 使用stat_ecdf 和geom_boxplot 函数分别绘制累积分布曲线图(图4C)与箱线图(图4D)以评估2 组样本中基因的整体表达水平。同样地,这2 种分析方法也都表明拟南芥根部的整体基因表达水平显著高于芽部。与图4A、B 一致,聚类图谱的结果同样表明组内重复的相关性较高,而组间差异较大(图4E)。同时,该结果还表明拟南芥根部的基因总体表达量高于芽部(图4E)。

图4 样本间基因表达量的可视化Fig.4 Visualization of gene expression levels among samples
2.4 差异基因的鉴定
RNA-seq 下游分析的第一步是鉴定样本间差异表达的基因。RNApipe 集成了当下主流的差异表达工具:DESeq2、edgeR、limma。这些工具具有不同的模型和优势(见Documentation.pdf),用户可以根据实验需求在config.yaml 中选择其中任一工具进行分析。RNApipe 默认使用DESeq2 软件在基因水平上执行差异基因的鉴定,DESeq2 使用基因表达的原始丰度而不是标准化丰度进行统计检验[28]。DESeq2分析的结果包括:对数变化(logFC)、错误发现率(FDR)以及调整后的P值。图5 展示了拟南芥芽部相较于根部显著差异基因(|logFC|≥1 且Padj<0.05)的数目,以及前30 个变化最大的显著差异基因的表达量热图。除了可视化图片,输出结果还包括基因表达矩阵和显著差异基因列表,以便科研人员进一步分析和后续验证。
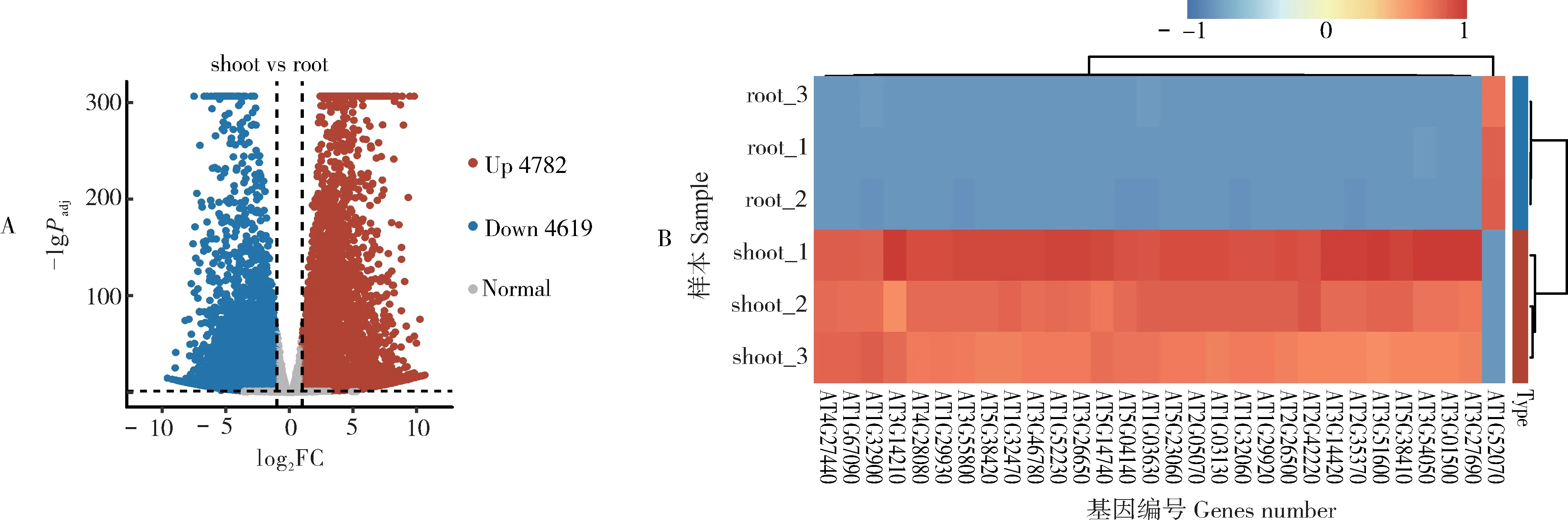
图5 样本间差异表达基因的可视化Fig.5 Visualization of differentially expressed genes among samples
2.5 功能注释
差异基因的功能注释对于了解特定条件所影响的生物学过程至关重要。基因本体论(GO)分析[29]与京都基因和基因组百科全书(KEGG)途径分析[30]是对差异表达基因进行分类的重要工具。RNApipe通过KOBAS-i 数据库对差异基因的生物学功能和参与的通路进行注释(图6)。GO 分析从分子功能(MF)、生物过程(BP)和细胞成分(CC)3 个方面展示,拟南芥根部与芽部间差异基因的GO 分类注释,表明差异基因主要与氧化还原和胁迫相应等过程相关,并显著富集于叶绿体、质体等部位(图6A)。KEGG 通路分析是探索差异基因的功能和相关通路的另一个重要工具。KEGG 气泡图展示了根部与芽部间差异基因显著富集的前30 条KEGG 通路,气泡的大小代表富集到该通路中基因数目的多少,气泡的颜色从红到蓝代表富集程度越来越显著。结果表明差异基因主要富集在代谢和植物激素信号转导等通路(图6B)。
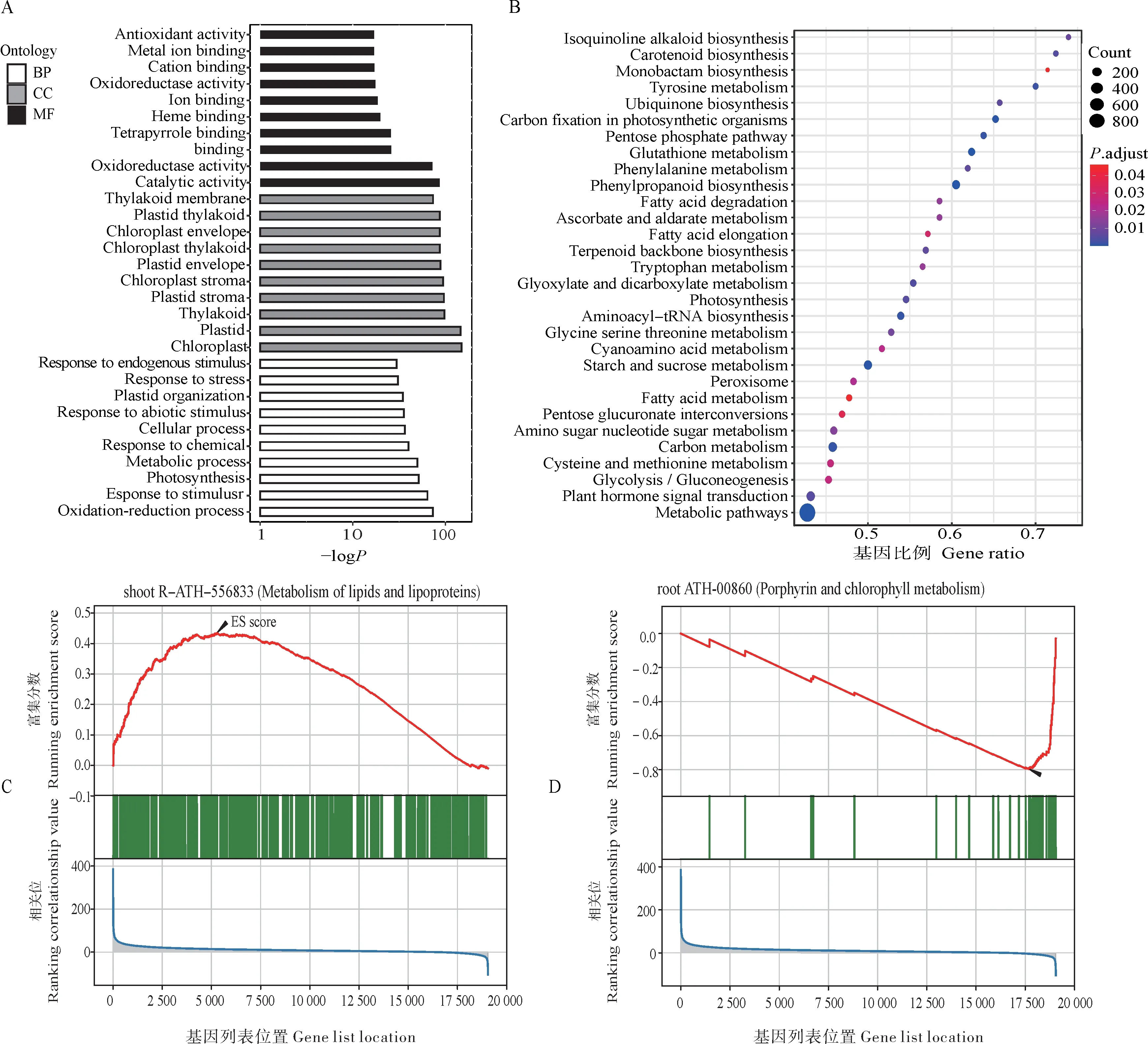
图6 样本间基因功能注释的可视化Fig.6 Visualization of gene functional annotations among samples
GO 和KEGG 富集分析往往侧重于识别两组间差异表达显著的基因,这可能导致遗漏部分差异表达不显著却有重要功能和意义的基因,另外,GO 和KEGG 分析无法判断差异基因的变化方向是上调还是下调。基因集富集分析(GSEA)不需要指定明确的差异阈值,算法会根据实际数据的整体趋势对基因的表达差异进行排序,随后检验数据库中预先设定的基因集富集在该排序列表的顶端或底端,因此,GSEA 检测到的是基因集而不是单个基因的表达变化。GSEA 图展示了脂质和脂蛋白通路中的基因集的表达在拟南芥的芽中呈现上调的趋势,而卟啉与叶绿素代谢通路中的基因集的表达在根中呈现下调的趋势(图6C)。
2.6 RNApipe与其他自动化分析工具的比较
与现有的自动化分析工具对比(表1),RNApipe通过Conda 虚拟环境保证了所有软件的自动安装与运行,通过Snakemake 工作流程管理系统整合各个分析步骤,保证了数据的可重复性与功能的可扩展性,并通过用户指定输入文件,保证了RNApipe 适用于所有有参物种的分析。先前的工作流程大多到鉴定差异基因就截止,然而差异基因的功能注释对于生物学问题的研究更具有指导意义,因此RNApipe通过KOBAS-i数据库进行功能注释。该数据库目前整合了最新的KEGG 数据库,支持5 944 个物种的KEGG 注释和71 个物种的GO 注释信息。另外,RNApipe 在各个分析步骤中集成了多款当下主流的软件,并在log 日志文件中记录同类型软件的运行情况。通过比较运行时间和所耗硬件资源,我们筛选出一套最优软件组合作为RNApipe 的默认流程(FastQC-fastp-HISAT2-featureCounts-DESeq2-KO⁃BAS-i)。同时,用户也可以根据自己的实验数据,通过修改config.yaml 中的参数选择非默认软件进行分析。RNApipe 另一重要的特点是在每一分析阶段都提供详细的高质量图表供用户直接使用。
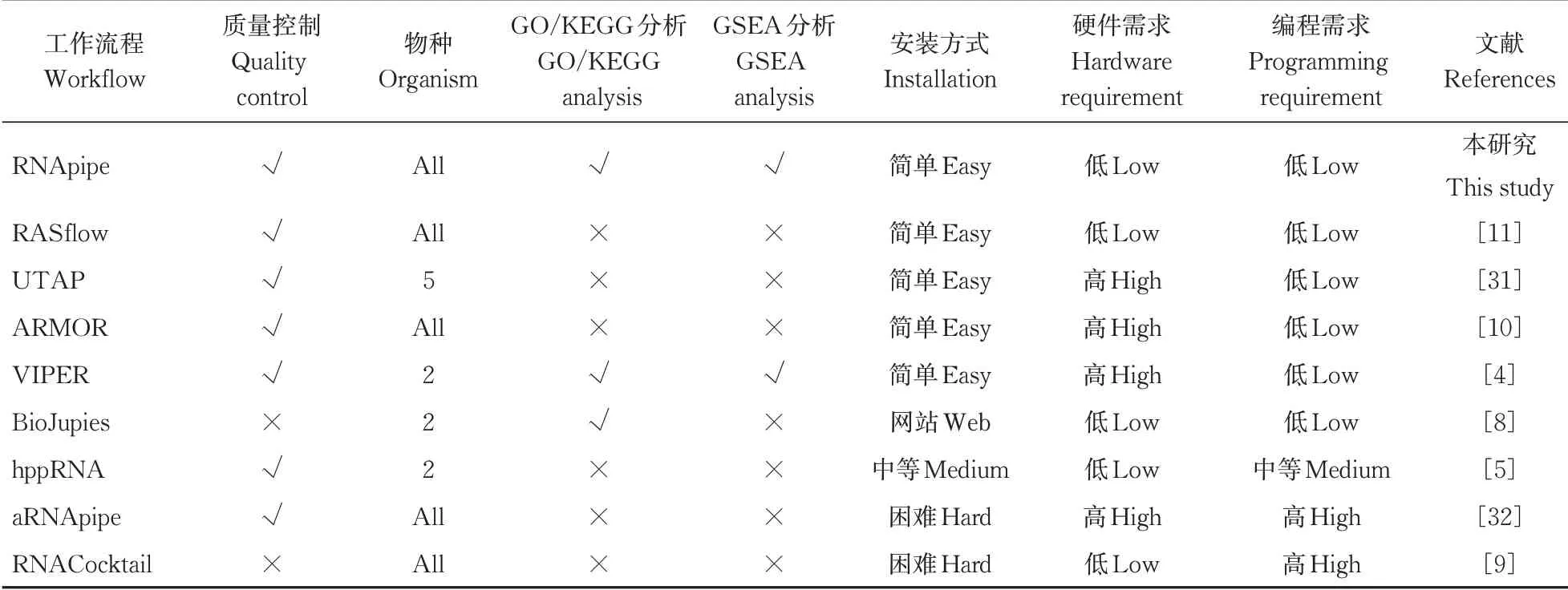
表1 RNApipe与其他自动化分析工具的比较Table 1 Comparison of RNApipe with other automated analysis tools
3 讨 论
利用RNA-seq 数据鉴定差异表达基因为研究重要的科学问题提供了思路,若将整个过程的分析步骤整合为一个自动化、模块化的流程,将极大地方便科研工作者的使用。为此,本研究开发了一个适用于任何有参物种的、流程更加完整的RNA-seq自动化分析工作流程:RNApipe。RNApipe工作流程旨在分析来自任何有参物种的RNA-seq数据。目前,RNApipe已在以下5个模式物种的完整数据集中完成测试与评估:人(PRJNA390636;GEO:GSE100075)、小 鼠(PRJNA630257;GEO:GSE149838)、拟南芥(PRJ⁃NA321304;GEO:GSE81332)、粗糙脉孢菌(PRJ⁃NA392079;GEO:GSE100539) 、酵 母(PRJ⁃NA318684;GEO:GSE80357)。分 析 结 果 表 明,RNApipe 运行稳定,功能注释结果符合已发表文献的数据。实际数据集的可视化结果存放于https://github.com/ywu019/RNApipe_pj.git.,与现有的自动化分析工具对比,RNApipe 具有安装容易、使用方便、易于扩展、功能丰富等特点。RNApipe 将帮助科研工作者以一种简单、高效且可重复的方式从RNAseq数据中获取有价值的信息。
RNApipe 目前主要适用于对二代Illumina 测序平台产生的短读长(short-read)数据进行差异基因分析。在RNApipe 的基础上,用户可以通过修改rules命令来扩展其他功能,例如可变剪切与单核苷酸变异的检测等。另外,由于常规RNA-seq 是对所有细胞的RNA 进行测序的技术,因此无法辨别细胞的类型和空间信息。近年来兴起的单细胞转录组测序技术(scRNA-seq)常应用于研究不同组织或器官中(例如正常和癌症)差异基因的表达,为研究单细胞水平的基因表达谱提供了机会。空间转录组测序技术通过将成像技术与scRNA-seq 相结合,可以绘制出转录本在组织中表达的位置,进一步提高组织分辨率。scRNA-seq 技术与空间转录组学技术的普及同时伴随着研究人员在数据分析方面面临的挑战,RNApipe 的结构设计则可以为scRNA-seq 自动化数据分析工具的构建提供参考。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
师道·教研(2022年1期)2022-03-12
中国生殖健康(2020年4期)2021-01-18
海洋信息技术与应用(2020年1期)2020-06-11
心电与循环(2020年1期)2020-02-27
传媒评论(2019年4期)2019-07-13
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
江苏农业科学(2017年5期)2017-04-15
