
In掺杂促进AuCuNi合金表面氧化膜形成的第一性原理计算
2023-01-09 12:00陈敬昶张慧蒙符荣刘学渊王远
表面技术 2022年12期
陈敬昶,张慧蒙,符荣,刘学渊,王远
In掺杂促进AuCuNi合金表面氧化膜形成的第一性原理计算
陈敬昶,张慧蒙,符荣,刘学渊,王远
(西南林业大学 机械与交通学院,昆明 650224)
采用第一性原理计算In掺杂促进AuCuNi合金表面氧化膜的形成机制,以为在金基合金中掺杂元素,促进表面氧化膜生成提供理论依据。基于构建适用于第一性原理计算、原子比Au:Cu:Ni=9:5:2的晶体结构模型,对In掺杂AuCuNi体系模型的稳定性、偏析特性以及吸附特性进行计算。In原子替代掺杂AuCuNi合金(111)面中各元素后形成新的AuCuNiIn表面,掺杂形成能均为负值,这说明In替位AuCuNi合金(111)面中任何一个原子都会促进AuCuNi表面的稳定性。当In替位掺杂AuCuNi表面第一层的Ni原子时,稳定性提升最大,掺杂形成能为–1.326 eV;当In替位掺杂AuCuNi表面第三层的Ni原子时,掺杂形成能最大为‒0.503 eV,这表明当In原子掺杂到该位置时,体系稳定性的提升最小。通过偏析能的计算发现,掺杂后的In有向其他位点偏析的趋势,最易向偏析能最小的位点偏析,即向表层Ni原子偏析,偏析能为‒0.739 eV。因此,使In原子替位掺杂第一层的Ni原子,形成最稳定的AuCuNiIn表面结构。此外,通过在AuCuNiIn表面吸附氧原子和计算吸附能发现,当原子顶位吸附时吸附能都比较高,这说明Au、Cu、Ni原子都不易在顶位吸附氧原子,其中3(Au)位点的吸附能为0.034 eV,其值大于0,说明Au原子的顶位不会自发地吸附氧原子。表层原子中吸附能最低的几个位点3(‒3.571 eV)、1(‒3.462 eV)、2(‒3.021 eV)的氧原子均与In原子成键,这说明In原子附近更易吸附氧原子。最后,通过电荷差分密度图和布居分析,发现O原子与周围其他原子有明显的电荷转移,并与In原子、Cu原子和Ni原子形成键。这进一步表明O原子与周围原子发生化学反应,提高了材料表面的稳定性,证实了吸附能计算的准确性。基于以上计算分析得出In原子的掺杂可以有效促进AuCuNi表面氧化膜的形成。通过第一性原理计算预测了元素掺杂对材料表面性能的影响,为掺杂促进材料表面氧化膜的形成提供了一定的理论参考。
金基合金;氧化膜;第一性原理计算;吸附特性;掺杂形成能
金基合金(AuNiCr、AuAgCu、AuCuNi)具有优异的化学惰性、导电性和耐腐蚀性等,已成为制造航空航天、精密电位器和电刷的主要材料[1-4]。金具有很强的化学稳定性,是高温下唯一不与氧发生反应的金属,金基合金中Au含量较多,表面难以形成致密的氧化膜,导致基体材料在高摩擦磨损环境中快速损耗[5],材料寿命大幅下降。所以在金基材料表面制备氧化膜具有重要意义。
为了促进基体材料表面氧化膜的生成,人们付出了巨大的努力。诸如,近年来微弧氧化技术制备材料氧化膜的方法被广泛应用[6-7]。唐艳茹等[8]采用微弧氧化技术在铝合金表面制备微弧氧化陶瓷薄膜,有效地提升了材料的耐磨性和耐蚀性。同时,学者们发现在金属表面掺杂其他物质也可以促进材料表面产生氧化膜[9]。虞澜等[10]发现在AuCuNi中分别加入Sm、Sc、Nd等稀土元素,在自然情况下材料表面能够与氧气反应形成氧化膜,从而提高材料的摩擦性能。根据何安莉等[11]对AuNiIn材料的研究发现,In在材料表面极易被氧化形成In2O3,且In2O3具有优异的力学性能和自润滑性能。事实上,这2种策略为金基材料表面氧化膜的制备提供了重要的参考依据。虽然微弧氧化工艺能在材料表面制备致密氧化膜,但是氧化膜质量受制于众多的工艺参数,难以精确控制[12]。同时,在AuCuNi中加入稀土中的元素虽能在材料表面产生氧化膜,但是其内在机理不清,限制了材料表面氧化膜的进一步调控制备。
值得注意的是,第一性原理计算方法为探究元素掺杂后材料改性机理提供了新的途径[13-15]。如Shi等[16]利用第一性原理计算了Au(111)表面氧化物形成的结构和稳定性,证明了氧化Au在纳米金催化剂的行为中可以发挥作用的观点。刘坤等[17-18]计算了NiTi(110)表面氧原子的吸附和扩散特性,结果表明,O原子在Ti上的吸附最稳定,揭示了NiTi合金表面产生TiO2的原因。这为探究元素掺杂后氧化膜产生的机理提供了一定参考。
基于以上分析,本文采用In掺杂AuCuNi,用第一性原理计算掺杂形成能、偏析能、吸附特性等,重点研究了In在AuCuNi中的赋存状态,探究In掺杂后基体材料表面氧化膜的形成机理。
1 计算方法及模型
1.1 计算方法
所有计算均是基于密度泛函理论(DFT)[19]的第一性原理的方法,使用MaterialsStudio软件中CASTEP程序包[20]进行。在广义梯度近似(GGA)[21]框架下,采用PW91泛函形式确定交换关联函数。Ni元素为磁性元素,为了消除磁矩、磁序或强交互作用对材料结构、能量的影响,本文在固态计算时打开了Spin polarized功能。采用超软赝势描述价电子与离子实之间的相互作用,平面波截断能为351 eV。自洽计算应用 Pulay混合密度法,收敛精度为2.0× 10−6eV/atom,在倒易空间中第一布里渊区点网格密度选取3×3×3。采用 BFGS算法对晶胞进行几何全优化(包括晶胞参数和原子内坐标),收敛条件为:体系能量小于2.0×10–5eV/atom,最大Hellmann-Feynman力低于0.5 eV/nm,原子最大偏移量小于0.000 2 nm,体系最大应力小于 0.1 GPa。
1.2 计算模型
由于AuCuNi是非化学计量结构[22],在研究In对AuCuNi表面掺杂之前,必须确定其稳定的结构。根据相关文献显示[23],原子比Au∶Cu∶Ni=9∶5∶2时,其结构较为稳定。因此,本文选择总原子数为32个,根据上述比例计算确定了原子比Au∶Cu∶Ni= 9∶5∶2的模型,如图1所示,晶格常数=0.815 6 nm,=90°。低指数晶面稳定性一般都强于较高指数晶面,容易形成稳定表面,所以本研究选取了AuCuNi结构的低指数晶面(111)为研究对象。由于不同截止端Au、Cu和Ni元素浓度差距较大,Au、Cu、Ni元素浓度最大相差分别为25.00%、12.50%和12.50%,因此需要计算各截止端表面的能量情况,以确定能量最低的截止端表面。共发现了6种首层原子浓度不同或原子排列顺序不同的截止端表面结构,图2a—f截止端首层原子比例Au∶Cu∶Ni分别为7∶6∶3、11∶4∶1、11∶4∶1、7∶6∶3、7∶6∶3、11∶4∶1。虽然截止端图2a、图2d、图2e首层原子浓度相同,截止端图2b、图2c、图2f首层原子浓度相同,但是其原子的分布位置各不相同,故而产生6种截止端。每个表面结构模型由4层原子层组成,每层16个原子,共64个原子,真空层设置为1.2 nm,晶格常数==1.153 5 nm,=2.206 3 nm,==90°,=60°。

图2 AuCuNi不同截止端表面结构模型
计算6种截止端的表面能,以确定不同截止端的稳定性,表面能由公式[24](1)给出。

式中:slab为纯净表面的总能;0为表面区域的面积(nm2);Au、Ni和Cu分别为体系中Au、Ni和Cu原子的个数;Au、Ni和Cu分别表示Au、Ni和Cu原子的化学势。
第一性原理计算所得不同截止端的表面能列于表1。由表1可知,表面能由大到小依次为截止端(d)1.019 J/m2>截止端(f)1.015 J/m2>截止端(c)1.009 J/m2> 截止端(a)1.007 J/m2>截止端(b)1.003 J/m2>截止端(e)0.984 J/m2,即截止端(e)的结构比其他截止端更加稳定,AuCuNi的(111)面更容易形成截止端(e)的表面结构。由于截止端(e)更加稳定更易形成,所以在后续的研究过程中以截止端(e)作为掺杂模型进行研究。

表1 不同截止端表面能
2 计算与分析
2.1 掺杂形成能计算
为了探究In原子掺杂后对AuCuNi合金表面氧化膜形成的影响,本文首先研究了In原子掺杂到AuCuNi合金后是否可以形成稳定的合金体系,进行了掺杂形成能的计算。掺杂分为替位掺杂和间隙掺杂2种,由于间隙掺杂需要掺杂原子半径较小,而In原子的半径较大,与Au、Cu、Ni原子半径相似符合替位掺杂的特点。故将In原子以替位的方式掺杂到AuCuNi低指数面最稳定的截止端(e)中,替位的位点如图3所示,其中图3a、图3b、图3c分别代表掺杂层数1、2、3,圈出位置代表Au、Cu、Ni在3层中被In替代的位置。

图3 掺杂位点示意图
根据公式(2)计算In取代不同原子的掺杂形成能。
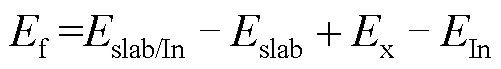
式中:slab/In为In掺杂后的总能量;slab为未掺杂In的总能量;x为被In置换的原子的能量;In为In原子的能量。
每次In原子只掺杂替位一层中的1个原子,掺杂后进行弛豫,故计算的掺杂形成能共有9种情况,列于表2,其中1、2、3代表掺杂的层数,Au、Cu、Ni代表被替代的原子类型。当掺杂形成能为负值时,说明界面稳定性比活性元素未掺杂前稳定性提升,当形成能为正值时,则说明界面稳定性较活性元素未掺杂时稳定性降低。据表2可知,掺In后,所有表面的掺杂形成能均为负值,说明In掺杂后的表面较In未掺杂时稳定,同时可以发现替位掺杂第一层的Ni原子掺杂形成能最小为‒1.326 eV,表明In最易取代掺杂到第一层的Ni原子上,形成掺杂后最稳定的表面,替位掺杂第一层的Au原子掺杂形成能最高为‒0.503 eV,说明当In原子掺杂到第一层Au原子位点时,AuCuNiIn合金体系稳定性最差。Zhang等[25]研究发现Au基合金中加入In元素能够有效改善合金的微观组织和提高材料的耐蚀性和硬度,说明In的掺杂能够提升Au基合金的稳定性,说明了掺杂形成能计算的准确性。通过各位点的掺杂形成能可以发现,当In原子替位掺杂同一层的原子时,替换Ni原子时的掺杂形成能最低。替位掺杂同类型原子时,随着原子层数的增加,Ni位点的掺杂形成能逐渐增加,Au位点的掺杂形成能是先下降后上升,Cu位点的掺杂形成能则是先上升后下降。同层之间不同位点的掺杂形成能最大相差0.823 eV,同种原子层数不同掺杂形成能最大相差0.552 eV,由于掺杂形成能相差较大,说明In原子掺杂后可能会产生偏析。而在In掺杂到GaAs[26]和TiO2[27]等体系的研究中,In原子均会向材料表面进行扩散偏析,这进一步说明In原子掺杂后会向表层偏析。
2.2 偏析能计算
为进一步探究In掺杂到AuCuNi中后是否会有向表面偏析的特性,本文对In的偏析能进行了计算,计算原理见公式(3)[28]。


表2 掺In在AuCuNi表面的掺杂形成能
In原子掺杂后的偏析能列于表3。偏析能表示某一位点原子向其他位点移动的趋势,偏析能越小说明移动的趋势最大。考虑到In原子掺杂3-Au位点的稳定性较差且3-Au位点在合金内部,因此将3-Au位点作为参照位点,比较其他掺杂位点(1-Cu、1-Ni和2-Au等)与3-Au的掺杂形成能,可以获得偏析能的大小。据表3可知,偏析能由小到大排序为1-Ni<1-Cu<2-Ni<3-Ni<2-Au<3-Cu<2-Cu<3-Au<1-Au,这说明In原子最易向1-Ni位点进行偏析,偏析能为‒0.739 eV,其次为1-Cu、2-Ni、3-Ni、2-Au、3-Cu、2-Cu、3-Au、1-Au等位点,这说明原子的偏析路径由1-Au依次经过3-Au、2-Cu、3-Cu、2-Au、3-Ni、2-Ni、1-Cu,最终到达1-Ni位置。同种原子随着原子层数的增加,偏析能逐渐增大,分别为1-Cu<3-Cu< 2-Cu、1-Ni<2-Ni<3-Ni、2-Au<3-Au<1-Au,同种原子易向第1、2层原子偏析,同层原子最易向Ni原子位点偏析,所以In掺杂后最易向第一层的Ni原子位点偏析。偏析能的计算结果与李波等[29]的试验结论(In掺杂到其他体系后会向表面进行偏析)一致。

表3 In原子的掺杂形成能和偏析能
2.3 表面吸附氧特性
2.3.1 吸附能计算
由于掺杂后的In原子易向表层偏析,为了探究偏析于AuCuNiIn表面的In能否形成氧化物,对In掺杂1-Ni位点(此时稳定性最好)后形成的AuCuNiIn表面进行了吸附O特性的分析,采用公式(4)进行吸附能的计算。

式中:slab/O为O原子吸附后的体系总能;slab为O原子未吸附时的纯净AuCuNiIn表面的能量,一个O2分子的能量为O2,则被吸附的O原子的能量为1/2O2。O原子吸附于AuCuNiIn表面,O原子共有18种不等价的吸附位点。其中1表示在Au、Ni、In和Cu原子的顶位吸附,为桥位吸附,为2种不等价的三角形孔隙位点。
O原子从不同的初始位置进行结构优化,优化后吸附点位如图4所示。可以发现,当初始吸附位置为桥位时,O原子会自发向三边形空隙位点移动,说明三边形吸附位点的稳定性较好。在Cu原子顶位吸附时,O原子自发地向桥位移动,说明桥位吸附的稳定性要优于顶位吸附。同时,当O原子吸附于Au原子上,经过优化后O原子会自发地吸附到其他原子上,说明Au原子不易与氧气反应或其他原子的活性高于Au原子。
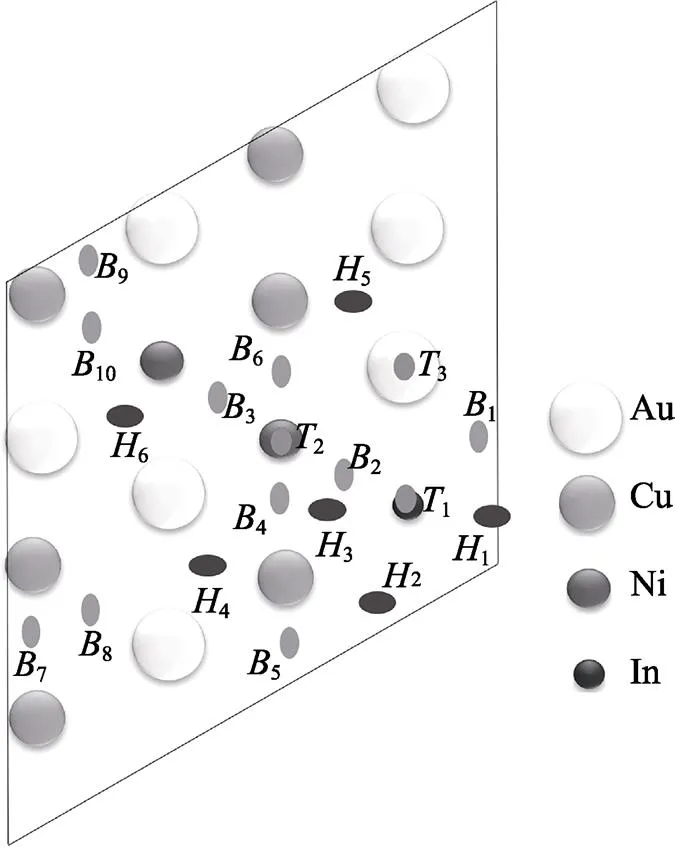
图4 O原子吸附位点示意图
不同位点吸附能计算结果见表4。吸附能小于0时,表明材料能够自发吸附,吸附能越小越容易吸附。对于AuCuNiIn表面,除3(Au原子顶位吸附)外所有吸附位点的吸附能均为负值,说明除Au原子顶位吸附外所有位点均能自发吸附氧原子。其中,孔位3的吸附能最小,为‒3.571 eV,表明O原子在该位点最易吸附。而当O原子吸附于Au原子的顶位(3)时,则具有最大的吸附能(0.034 eV),表明此位置最不易吸附O原子且不会自发吸附。由图4可以看出,吸附能低于‒3 eV的3个位点中,最低的2个为空位吸附,分别为3位点(‒3.571 eV)和1位点(‒3.462 eV),然后为桥位吸附2位点(‒3.021 eV)。顶位的吸附能都比较大,分别为1(‒0.526 eV)、2(‒1.659 eV)、3(0.034 eV)。通过观察1、2和2的位置可以看出,吸附能最低的3个位置中都在In原子的周围,且O原子在1、2和2位点时都与In原子成键,说明In原子掺杂后促进了AuCuNi合金表面与氧气结合的能力。高攀等[30]在AgSn中掺杂In的试验中发现,In的添加有利于促进合金表面氧化膜的形成,证明了第一性原理计算In掺杂AuCuNi能促进表面氧化膜形成的可行性。
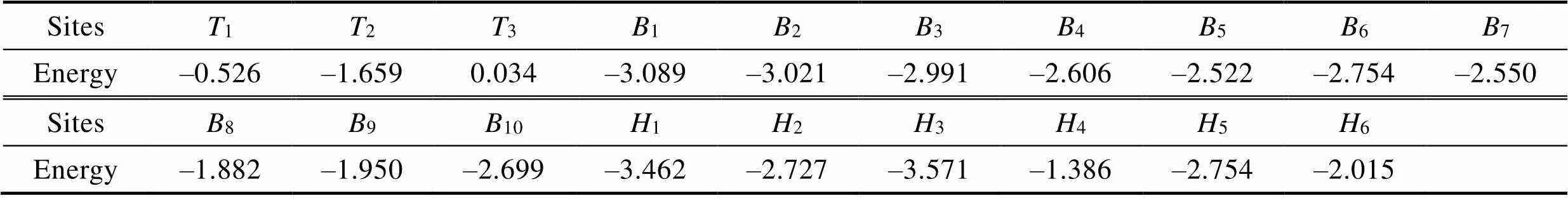
表4 氧原子吸附于不等价位点的吸附能
2.3.2 电子结构分析
为了进一步观察O原子的成键情况,从电子角度对O原子在3位点的表面进行了电荷差分密度分析和布居分析。电荷差密度来描述成键前后原子电荷密度的差异,电荷的积累和耗散可以直接反映化学键的增强和减弱。通过图5可以发现,氧原子发生了明显的电荷累计,说明氧原子参与了与周围其他原子的化学成键,提高了表面的稳定性,这与3位点时吸附能计算结果一致。通过电荷布居分析能够了解各原子轨道上的分布和电荷转移,进而确定原子间的成键情况。电荷布居分析发现O原子的charge为负值,等于‒0.67 eV,这说明O原子得到了较多的电荷,表现出较强的共价性。通过键布居分析发现O(1)原子分别与Ni(3)原子、Cu(18)原子、In(1)原子成键,键布居数分别为0.52、0.47、0.35,键布居数都为正值这说明O原子与Ni、Cu、In原子成键都为共价键。从电子角度解释了O原子在表面吸附的内在机理,与吸附能计算结果一致。

图5 差分电荷密度图
3 结论
1)In原子替代掺杂AuCuNi合金(111)面中各元素后形成新的AuCuNiIn表面,掺杂形成能均为负值,表明In原子掺杂后有助于提升材料的稳定性。通过偏析能的计算发现,In原子掺杂后更易向表面析出且最易析出到第一层的Ni原子位点形成更稳定的结构。
2)AuCuNiIn表面吸附O特性的计算发现,吸附能低于‒3 eV的3种位置中,O原子都与In原子有键连接,说明AuCuNi合金中掺杂In原子会促进表面氧化膜的形成。电荷差分密度分析和布居分析,通过电荷密度差异和电子转移情况,从电子角度证明了O原子与In原子成共价键连接,这与吸附能计算结果一致。
[1] 李镇隆, 梁江, 王肖铮, 等. 自润滑电接触材料与电刷滑环元件[J]. 电工材料, 2008(1): 25-27.
LI Zhen-long, LIANG Jiang, WANG Xiao-zheng, et al. Self-Lubricating Electric Contact Material and Brush Slip Ring Element[J]. Electrical Engineering Materials, 2008(1): 25-27.
[2] 刘义, 王惠, 高兴珍. 航空用几种贵金属合金线绕电位器材料耐磨特性研究[J]. 仪表材料, 1984, 15(3): 8-13.
LIU Yi, WANG Hui, GAO Xing-zhen. Study on Wear Resistance of Wire-Wound Potentiometer Materials with Several Precious Metal Alloys for Aviation[J]. Journal of Functional Materials, 1984, 15(3): 8-13.
[3] 官达高. 电位器用自润滑贵金属合金初探[J]. 仪表材料, 1980, 11(6): 69.
GUAN Da-gao. Preliminary Study on Self-Lubricating Precious Metal Alloy with Potentiometer[J]. Journal of Functional Materials, 1980, 11(6): 69.
[4] 毕珺, 张昆华, 耿永红, 等. 金基六元合金电接触材料的显微组织和性能研究[J]. 贵金属, 2011, 32(3): 31-35.
BI Jun, ZHANG Kun-hua, GENG Yong-hong, et al. Microstructure and Mechanical Properties of Au-Base Alloy Electrical Contact Materials[J]. Precious Metals, 2011, 32(3): 31-35.
[5] ANTLER M. Sliding Wear of Metallic Contacts[J]. IEEE Transactions on Components, Hybrids, and Manufacturing Technology, 1981, 4(1): 15-29.
[6] 赵灏琳, 赵李斌. AZ31B镁合金挤压板表面在酸性环境下自愈合微弧氧化膜的制备与判断研究[J]. 中国设备工程, 2020(S2): 133-138.
ZHAO Hao-lin, ZHAO Li-bin. Preparation and Judgment of Self-Healing Micro-Arc Oxidation Film on AZ31B Magnesium Alloy Extruded Plate in Acid Environment[J]. China Plant Engineering, 2020(S2): 133-138.
[7] 肖鹏, 冯坤, 孙瑞, 等. 新型医用镁合金微弧氧化膜层制备及性能评价[J]. 科技与创新, 2020(24): 1-4.
XIAO Peng, FENG Kun, SUN Rui, et al. Preparation and Performance Evaluation of Micro-Arc Oxidation Coating on Medical Magnesium Alloy[J]. Science and Technology & Innovation, 2020(24): 1-4.
[8] 唐艳茹, 潘利华, 常宇, 等. 微弧氧化提高铝合金耐磨性能的研究[J]. 表面技术, 2015, 44(2): 48-54.
TANG Yan-ru, PAN Li-hua, CHANG Yu, et al. Improving the Wear Resistance of Aluminum Alloy through the Micro-Arc Oxidation Technology[J]. Surface Technology, 2015, 44(2): 48-54.
[9] 李伟. Re改性铝化物涂层的制备和氧化行为研究[D]. 合肥: 中国科学技术大学, 2021.
LI Wei. Preparation and Oxidation Behaviors of Rhenium Doped Aluminide Coatings[D]. Hefei: University of Science and Technology of China, 2021.
[10] 虞澜, 冯本政. Au-Cu-Ni-RE合金摩擦副磨损特性研究[J]. 贵金属, 1999, 20(4): 7-11.
YU Lan, FENG Ben-zheng. The Study on Friction and Wear Behaviour of Au-Cu-Ni-RE Alloys Sliding Pairs[J]. Precious Metals, 1999, 20(4): 7-11.
[11] 何安莉, 陶春虎, 郭效东, 等. 含铟电刷材料自润滑耐磨机理的研究[J]. 航空材料学报, 1989, 9(3): 1-8.
HE An-li, TAO Chun-hu, GUO Xiao-dong, et al. The Study of Self-Lubricating Wearresistant Mechanism in indium-Containing electro-Brush Alloy[J]. Journal of Aeronautical Materials, 1989, 9(3): 1-8.
[12] 王亚明. Ti6Al4V合金微弧氧化涂层的形成机制与摩擦学行为[D]. 哈尔滨: 哈尔滨工业大学, 2006.
WANG Ya-ming. Formmation Mechanism and Tribological Behavior of Microarc Oxidation Coatings on Ti6Al4V Alloy[D]. Harbin: Harbin Institute of Technology, 2006.
[13] 毛斐, 吕皓, 唐法威, 等. Mn和In添加对SmCo7结构稳定性及磁矩影响的第一性原理计算[J]. 金属学报, 2021, 57(7): 948-958.
MAO Fei, LU Hao, TANG Fa-wei, et al. First-Principle Calculation on the Effect of Mn and in on the Structural Stability and Magnetic Moment of SmCo7Alloys[J]. Acta Metallurgica Sinica, 2021, 57(7): 948-958.
[14] ZHANG Ying, WANG Jing-qin, KANG Hui-ling. Study on Electrical Properties of AgSnO2Contact Materials Doped with Rare-Earth La, Ce, and Y[J]. IEEE Transactionson Components, Packaging and Manufacturing Technology, 2019, 9(5): 864-870.
[15] 雷博程, 夏桐, 彭彩云, 等. 稀土元素(Nd/Sm/Gd/Dy)掺杂SnO2的第一性原理计算[J]. 人工晶体学报, 2018, 47(8): 1625-1631.
LEI Bo-cheng, XIA Tong, PENG Cai-yun, et al. First- Principles Study on SnO2Doped Rare Earth Elements (Nd/Sm/Gd/Dy)[J]. Journal of Synthetic Crystals, 2018, 47(8): 1625-1631.
[16] SHI Hong-qing, STAMPFL C. First-Principles Investigations of the Structure and Stability of Oxygen Adsorption and Surface Oxide Formation at Au(111)[J]. Physical Review B, 2007, 76(7): 075327.
[17] 刘坤, 王福合, 尚家香. NiTi(110)表面氧原子吸附的第一性原理研究[J]. 物理学报, 2017, 66(21): 216801.
LIU Kun, WANG Fu-he, SHANG Jia-xiang. First-PrinciplesStudy on the Adsorption of Oxygen at NiTi(110)surface[J]. Acta Physica Sinica, 2017, 66(21): 216801.
[18] 刘坤, 王福合. NiTi(110)表面氧原子吸附和扩散的第一性原理研究[J]. 材料保护, 2016, 49(S1): 65-67.
LIU Kun, WANG Fu-he. First-Principles Study on Adsorption and Diffusion of Oxygen Atoms on NiTi(110) Surface[J]. Materials Protection, 2016, 49(S1): 65-67.
[19] KOHN W, SHAM L J. Self-Consistent Equations Including Exchange and Correlation Effects[J]. Physical Review, 1965, 140(4A): A1133-A1138.
[20] CLARK S J, SEGALL M D, PICKARD C J, et al. First Principles Methods Using CASTEP[J]. Zeitschrift Für Kristallographie-Crystalline Materials, 2005, 220(5-6): 567-570.
[21] PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation[J]. Physical Review B, Condensed Matter, 1992, 46(11): 6671-6687.
[22] KOGACHI M. Axial Ratio Change in the Ternary AuCu1–yNi, AuCu1–yPdand AuCu1–yPtSystems with Ll0-Type Structure[J]. Transactions of the Japan Institute of Metals, 1985, 26(3): 153-159.
[23] 袁晓虹, 李伟, 刘俊峰, 等. 时效处理对AuCuNi合金的微观结构及力学性能的影响[J]. 贵金属, 2020, 41(2): 21-24.
YUAN Xiao-hong, LI Wei, LIU Jun-feng, et al. Effects of Aging Treatment on the Microstructure and Mechanical Properties of AuCuNi Alloy[J]. Precious Metals, 2020, 41(2): 21-24.
[24] PENG Cheng, et al. Influence of Au, Cu, Pd Added in Ag Alloy on Stability and Electronic Structure of Ag/Al Interface by First-Principles Calculations[J]. Materials Today Communications, 2020, 22: 100670.
[25] ZHANG Jun-kai, et al. Properties and Microstructure Changes in Au-Cu-Based Alloy with Indium Addition[J]. Journal of Alloys and Compounds, 2018, 734: 81-88.
[26] MARTINI S, QUIVY A A, LAMAS T E, et al. Real-Time RHEED Investigation of Indium Segregation in InGaAs Layers Grown on Vicinal GaAs(001) Substrates[J]. Physical Review B, 2005, 72(15): 153304.
[27] ATANACIO A J, BAK T, NOWOTNY J. Effect of Indium Segregation on the Surface Versus Bulk Chemistry for Indium-Doped TiO2[J]. ACS Applied Materials & Interfaces, 2012, 4(12): 6626-6634.
[28] 徐沛瑶, 王宇飞, 高海燕, 等. 合金元素对Cu/γ-Fe界面特性影响的第一性原理研究[J]. 中国有色金属学报, 2018, 28(1): 39-45.
XU Pei-yao, WANG Yu-fei, GAO Hai-yan, et al. First- Principles Study of Effects of Alloying Elements on Cu/γ-Fe Interfacial Properties[J]. The Chinese Journal of Nonferrous Metals, 2018, 28(1): 39-45.
[29] 李波, 刘心宇, 黄锡文, 等. 微量添加In对Ag-Sn合金粉末氧化机理的影响[J]. 稀有金属材料与工程, 2014, 43(8): 1964-1968.
LI Bo, LIU Xin-yu, HUANG Xi-wen, et al. Effects of Minor in Addition on Oxidation Mechanism of Ag-Sn Alloy Powders[J]. Rare Metal Materials and Engineering, 2014, 43(8): 1964-1968.
[30] 高攀, 刘心宇, 李波. Ag-Sn-In合金粉末的氧化参数[J]. 稀有金属材料与工程, 2017, 46(10): 3023-3027.
GAO Pan, LIU Xin-yu, LI Bo. Oxidation Parameters of Ag-Sn-in Alloy Powders[J]. Rare Metal Materials and Engineering, 2017, 46(10): 3023-3027.
First-principles Calculation of Promoting the Formation of Oxide Film on AuCuNi Alloy Surface by In Doping
(School of Mechanical Engineering and Transportation, Southwest Forestry University, Kunming 650224, China)
Au is difficultly satisfied with work in the extreme environments such as high friction, high wear and strong corrosion due to it hardly reacts with oxygen to form a dense oxide film. Therefore, the formation mechanism of In doping in AuCuNi alloy to form the surface oxide film was calculated by First-principles in order to provide a theoretical basis for promoting the formation of surface oxide film by doping elements in the Au-based alloy in this paper. A crystal structure model with Au:Cu:Ni=9:5:2 suitable for First-principles calculations was constructed to calculate the stability, bias properties and adsorption properties of AuCuNi system with In doping. The results show: the doping formation energies of all new forming AuCuNiIn surfaces with In doping substitution for each element in AuCuNi (111) face are negative, this indicates that the stability of the AuCuNi surface can be promote by In substitution for any element in the AuCuNi (111) face, and when In substitution Ni atom in the first layer on AuCuNi surface, the greatest stability enhancement occurs with a doping formation energy of ‒1.326 eV. The maximum doping formation energy is ‒0.503 eV when In atoms substituting for Ni atoms in the third layer, which indicates that the increasing of the stability of the system is minimum at In atoms locating on the site. The bias energy calculation indicates that the doped In has a tendency to bias towards other sites and most easily towards the site of Ni atom with the lowest bias energy with value of ‒0.739 eV so the most stable AuCuNiIn surface structure is obtained when In substitute for a Ni atoms on the first layer. In addition, it is found that the adsorption energies are relatively high when In is adsorbed in the top position by the calculation of the adsorption energies, this indicated that Au, Cu and Ni atoms aren’t prone to adsorbing oxygen atoms in the top position, as well as the adsorption energy at3(Au) site is positive with value of 0.034 eV to explain Au on the top site don’t spontaneously adsorb oxygen atoms. All the oxygen atoms at the lowest adsorption energies in the surface atoms,3(‒3.571 eV),1(‒3.462 eV) and2(‒3.021 eV) are bonded to In atom to suggest that oxygen atom is more readily adsorbed near In atoms. Finally, it is found that O atom obviously transfers charge with the other surrounding atoms, and forms bonds with In, Cu and Ni atom by the charge differential density map and population analysis, i.e., a chemical reaction occurs between the O atom and the surrounding atoms, which improves the stability of the material and confirms the accuracy of the adsorption energy calculation. Based on the above calculation and analysis, it is concluded that the formation of oxide films on the AuCuNi surface can be effectively promote by In doping. In addition, the effects of doping element on surface properties were predicted by first-principles calculations, which provided a certain theoretical reference for promoting the formation of oxide films by doping element in the surface of materials in this paper.
gold-base alloy; oxide film; first-principles calculation; adsorption characteristics; doping formation energy
Tb43
A
1001-3660(2022)12-0101-08
10.16490/j.cnki.issn.1001-3660.2022.12.009
2022–08–09;
2022–12–06
2022-08-09;
2022-12-06
云南省农业基础研究联合专项基金(2018FG001-062);国家自然科学基金(51301144);云南省教育厅科学研究基金项目(2022J0500)
Joint Special Fund for Agricultural Basic Research of Yunnan Province (2018FG001-062) and National Natural Science Foundation of China (51301144); Scientific Research Fund Project of Education Department of Yunnan Province (2022J0500)
陈敬昶(1996—),男,硕士研究生,主要研究方向为材料表面改性。
CHEN Jing-chang (1996-), Male, Master, Research focus: material surface modification.
刘学渊(1979—),男,高级实验师,主要研究方向为计算材料与汽车尾气排放。
LIU Xue-yuan(1979-), Male, Senior laboratory technician, Research focus: computational materials and vehicle exhaust emission.
陈敬昶, 张慧蒙, 符荣, 等. In掺杂促进AuCuNi合金表面氧化膜形成的第一性原理计算[J]. 表面技术, 2022, 51(12): 101-108.
CHEN Jing-chang, ZHANG Hui-meng, FU Rong, et al. First-principles Calculation of Promoting the Formation of Oxide Film on AuCuNi Alloy Surface by In Doping[J]. Surface Technology, 2022, 51(12): 101-108.
责任编辑:万长清
猜你喜欢
教育家(2022年19期)2022-05-13
物理学报(2022年8期)2022-04-27
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
成都信息工程大学学报(2021年2期)2021-07-22
鞍钢技术(2021年2期)2021-04-20
河南冶金(2020年3期)2020-09-10
华东师范大学学报(自然科学版)(2020年1期)2020-03-16
数码设计(2017年4期)2017-11-01
