
Do tau-synaptic long-term depression interactions in the hippocampus play a pivotal role in the progression of Alzheimer’s disease?
2023-01-21 04:42ZhengtaoHuTomasOndrejcakPengpengYuYangyangZhangYinYangIgorKlyubinSeanKennellyMichaelRowanNengWeiHu
中国神经再生研究(英文版) 2023年6期
Zhengtao Hu,Tomas Ondrejcak,Pengpeng Yu,Yangyang Zhang,Yin Yang,,Igor Klyubin,Sean P.Kennelly,Michael J.Rowan,Neng-Wei Hu,,
Abstract Cognitive decline in Alzheimer’s disease correlates with the extent of tau pathology,in particular tau hyperphosphorylation that initially appears in the transentorhinal and related regions of the brain including the hippocampus.Recent evidence indicates that tau hyperphosphorylation caused by either amyloid-β or long-term depression,a form of synaptic weakening involved in learning and memory,share similar mechanisms.Studies from our group and others demonstrate that long-term depression-inducing low-frequency stimulation triggers tau phosphorylation at different residues in the hippocampus under different experimental conditions including aging.Conversely,certain forms of long-term depression at hippocampal glutamatergic synapses require endogenous tau,in particular,phosphorylation at residue Ser396.Elucidating the exact mechanisms of interaction between tau and long-term depression may help our understanding of the physiological and pathological functions of tau/tau (hyper)phosphorylation.We first summarize experimental evidence regarding tau-longterm depression interactions,followed by a discussion of possible mechanisms by which this interplay may influence the pathogenesis of Alzheimer’s disease.Finally,we conclude with some thoughts and perspectives on future research about these interactions.
Key Words: aging;Alzheimer’s disease;amyloid-β;Aβ oligomers;hippocampus;long-term depression;long-term potentiation;LTD;LTP;metabotropic glutamate receptor;N-methyl-D-aspartate receptor;tau hyperphosphorylation;tau phosphorylation;tau
From the Contents
Introduction 1213
Search Strategy and Selection Criteria 1214
Physiological Functions of Tau/Tau Phosphorylation 1214
Tau Hyperphosphorylation in Alzheimer’s Disease 1214
The Role of Tau in Hippocampal Long-Term Depression 1214
Hippocampal Long-Term Depression Promotes Tau Phosphorylation 1215
Conclusion and Future Research 1216
Introduction
Alzheimer’s disease (AD),the most common form of dementia in the elderly population,is characterized by an age-associated progressive cognitive decline (Scheltens et al.,2021).The neuropathological hallmarks of AD include amyloid-β (Aβ) accumulation in amyloid plaques,tau aggregation in neurofibrillary tangles,and brain atrophy caused by loss of synapses and neurons (Henstridge et al.,2019).Although both Aβ and tau are causally linked to cognitive impairment in patients with AD,clinical evidence indicates that the progressive cognitive decline correlates strongly with the extent of tau pathology,in particular the degree and nature of tau phosphorylation(Wegmann et al.,2019;Wesseling et al.,2020;Nies et al.,2021).Although tau is predominantly an intracellular protein,many forms of tau are present in the interstitial fluid,cerebrospinal fluid (CSF),and plasma.Aberrant elevation of CSF or plasma levels of phosphorylated tau (p-tau) at residues Thr181(p-tau181),Thr217 (p-tau217),and Thr231 (p-tau231) were recently proposed to be particularly sensitive markers of preclinical AD,long before clinical symptoms of dementia occur (Hansson,2021;Zetterberg and Blennow,2021;Karikari et al.,2022).
Synapses are known to be particularly susceptible to disruption by rogue forms of Aβ and tau.Recent findings confirm a significant association between synaptic loss in the neocortex and limbic system and the progression of cognitive impairment in patients with AD (Colom-Cadena et al.,2020;Mecca et al.,2020,2022;Nakamura et al.,2021).
The normal function,structure,and number of synapses change dynamically over time in an activity-dependent manner,forms of synaptic plasticity which are crucial for cognitive functions (Magee and Grienberger,2020).The most studied forms of synaptic plasticity,long-term potentiation (LTP) and longterm depression (LTD),are characterized by persistent strengthening and weakening of synaptic connectivity,respectively.LTP and LTD in animals,and LTP/LTD-like plasticity in humans,can be induced naturalistically without artificial electrical stimulation in brain areas including primary sensory and motor cortices (Whitlock et al.,2006;Cooke and Bear,2014;Mansvelder et al.,2019;Lengali et al.,2021).Synaptic LTP/LTD is engaged during cellular“engram” (memory trace) encoding during neuronal activity associated with different forms of learning (Magee and Grienberger,2020).In rodent hippocampus,LTP is often induced by high-frequency stimulation protocols typically involving delivery of one or several trains of stimuli at 100–400 Hz for 1 second.This,and/or similar high-frequency stimulation protocols trigger a strong transient summation of the excitatory postsynaptic potentials.The temporal large depolarization of postsynaptic cells is sufficient to relieve the magnesium block of the N-methyl-D-aspartate receptors (NMDAR) and results in a large amount of calcium entering the postsynaptic cells in a short period.In contrast,LTD is commonly induced by low-frequency stimulation(LFS) protocols that typically involve stimulation at 1–3 Hz for 5–15 minutes.This triggers a modest postsynaptic depolarization,resulting in a modest but prolonged calcium flux into the postsynaptic cells.The regulated trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs)to and from synaptic membranes is a key mechanism/component underlying synaptic LTP and LTD expression in many circuits;AMPARs are inserted into the postsynaptic membrane during LTP,while endocytosis pathways mediate the removal of AMPARs from the synapses (Figure 1).On the one hand,both LTP and LTD are used to study the neurophysiological substrates of normal learning and memory function (Connor and Wang,2016;Magee and Grienberger,2020),while on the other hand,disruption of both processes at hippocampal glutamatergic synapses are posited to be early events in AD(Benarroch,2018;Styr and Slutsky,2018;Chen et al.,2019;Li and Selkoe,2020;Padmanabhan et al.,2021).Interestingly,recent studies indicate that the presumed “physiological” electrical LFS of afferent fibers drives tau phosphorylation at Ser396 (p-tau396) in the hippocampus (Kimura et al.,2014;Regan et al.,2015;Suer et al.,2021),thereby enabling endogenous tau to promote “physiological” LTD (Kimura et al.,2014;Regan et al.,2015).Taylor et al.(2021) very recently discovered that chronic application of either LTD-inducing LFS or Aβ oligomers causes tau hyperphosphorylation via similar mechanisms in organotypic hippocampal slices.Aging is the most important risk factor for AD (Knopman et al.,2021).Kimura et al.(2017) found that LTDinducing strong LFS increases p-tau396 in the hippocampus of aged mice and we very recently reported that LTD-inducing standard LFS enhances p-tau181 and p-tau217 in the hippocampus of live aged rats (Zhang et al.,2022b).Taken together,the above evidence from our group and others suggests that tau-LTD interactions in the hippocampus may play a pivotal role in the pathological process of AD.
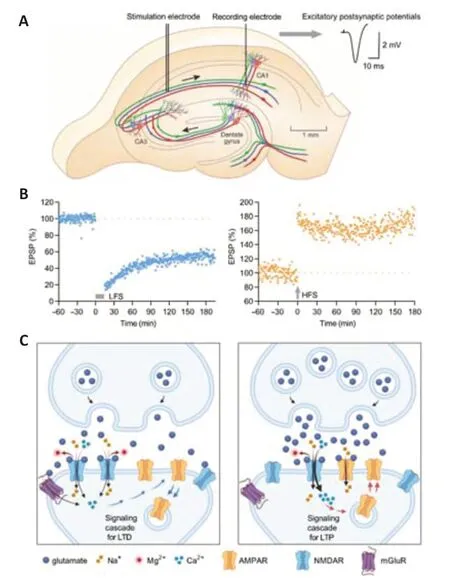
Figure 1|Schematic diagram of long-term potentiation and long-term depression at CA3-CA1 synapses in the hippocampus.
Search Strategy and Selection Criteria
The search strategy and selection criteria were limited to articles published in peer-reviewed journals.We searched the National Library of Medicine(PubMed) and Web of Science from inception to June 2022 using the following keywords: Alzheimer’s disease,tau phosphorylation,tau hyperphosphorylation,amyloid-β,Aβ oligomers,LTD,LTP,hippocampus,aging.A manual search of references of all retrieved studies,relevant reviews,and systematic reviews was also carried out.Studies were cited based on their relevance to the review.
Physiological Functions of Tau/Tau Phosphorylation
Tau,a microtubule-associated protein,is distributed abundantly in the central nervous system with a role in regulating neuronal microtubules and promoting axonal outgrowth.Qiang et al.(2018) reported that tau,rather than directly stabilizing axonal microtubules,enables them to have long labile domains.Recent evidence reveals more physiological functions including myelination,glucose metabolism,axonal transport,microtubule dynamics,iron homeostasis,neurogenesis,motor function,and DNA protection (Kent et al.,2020).
Tau protein contains 85 potential phosphorylation residues including 45 serine,35 threonine,and 5 tyrosine sites (Drepper et al.,2020).Phosphorylation of tau is the most intensively studied among posttranslational tau modifications.Although it is still not well understood,tau phosphorylation has been observed in physiological conditions such as brain development(Hefti et al.,2019),hypothermia (Bretteville et al.,2012),and hibernation(Arendt et al.,2003).
Tau Hyperphosphorylation in Alzheimer’s Disease
Different pathological changes of tau lead to different clinical phenotypes,manifesting as AD,frontotemporal dementia (including Pick’s disease as a sub-type),corticobasal degeneration,and progressive supranuclear palsy,collectively termed tauopathies (Simic et al.,2016;Zhang et al.,2022a).Apart from these adult-onset tauopathies,upregulated levels of abnormal tau are also found in certain neurodevelopmental disorders in children(Rankovic and Zweckstetter,2019).In AD,studies on tau mainly focus on posttranslational modifications (Wesseling et al.,2020) including but not limited to hyperphosphorylation (Grundke-Iqbal et al.,1986),truncation (Novak,1994;Zilka et al.,2006;Amadoro et al.,2020),ubiquitination (Petrucelli et al.,2004),glycation (Yan et al.,1994),glycosylation (Wang et al.,1996),nitration(Horiguchi et al.,2003),acetylation (Tracy et al.,2016),and sumoylation (Princz and Tavernarakis,2020).
Under pathological conditions of AD,tau is hyperphosphorylated at multiple residues such as Ser202/Thr205,Thr212,Ser214,Thr217,Ser262,and Ser422 (Miao et al.,2019).Hyperphosphorylated tau decreases microtubule binding which results in loss of axonal integrity (Qiang et al.,2018).Hyperphosphorylated tau also migrates from axons into the somatodendritic compartment,which increases Fyn,a 59 kDa member of the Src family of tyrosine kinases,in dendritic spines.High levels of Fyn lead to intracellular Ca2+flooding by phosphorylating postsynaptic GluN2B-containing NMDAR.Aberrant elevation of intracellular Ca2+induces excitotoxicity and eventually causes neuronal death (Chen et al.,2019;Barron et al.,2020).
Although tau mainly exists in the intracellular compartment,relatively high levels of both total tau and p-tau are also present in CSF of AD patients(Brier et al.,2016).Elevated CSF total tau levels in AD have been associated with axonal loss (Bos et al.,2019).Recent findings indicate that neurons can actively generate and secrete tau into the extracellular space in the brains of AD patients and healthy people (Sato et al.,2018;Ebashi et al.,2019).Sato et al.(2018) also observed the process of generating and releasing tau from differentiated neurons derived from human induced pluripotent stem cells and found that tau started to appear in the growth medium 3 days after the neurons were cultured.We recently reported that tau species in the secretomes of human induced pluripotent stem cell-derived neuronal models of early-onset AD also disrupt hippocampal LTP after acute injection in the rodent brain (Hu et al.,2018).Other recent studies also found that hyperphosphorylated and/or oligomeric tau can spread through transsynaptic transmission (Takeda et al.,2015;Gibbons et al.,2019;Jadhav et al.,2019;Pernegre et al.,2019).As noted in the introduction,increases in CSF or plasma levels of p-tau181,p-tau217,and p-tau231 are proposed to be particularly sensitive markers of preclinical AD,long before clinical symptoms of dementia(Hansson,2021;Zetterberg and Blennow,2021;Karikari et al.,2022).
In addition to being a promising biomarker for tracking early AD,tau phosphorylation has also become an attractive therapeutic target (Wegmann et al.,2021;Xia et al.,2021).Several recently completed or ongoing clinical trials focus on therapeutic strategies to decrease tau phosphorylation,including kinase inhibitors,phosphatase activators,and tau immunotherapy targeting specific p-tau species.Moreover,phosphorylation of tau is emerging as an important posttranslational modification relevant to both its physiological and pathological functions,making it urgent to identify specific modifications as therapeutic targets (Robbins et al.,2021;Wegmann et al.,2021;Xia et al.,2021).Different from physiological tau phosphorylation,which is generally considered reversible,hyperphosphorylated tau irreversibly self-assembles into toxic amorphous aggregates (Meng et al.,2022).
In summary,tau hyperphosphorylation leads to tau aggregation and correlates strongly with progressive cognitive decline in patients with AD.Although traditionally associated with microtubule dysfunction hyperphosphorylated tau also promotes synaptic disruption,a likely proximate cause of cognitive impairment in AD.The recent development of new assays enables the measurement of minute amounts of p-tau species in both CSF and plasma.Increased levels of p-tau181,p-tau217,and p-tau231 are particularly sensitive markers of preclinical AD.Therefore,tau immunotherapy targeting these p-tau species may prove beneficial very early in the disease.
The Role of Tau in Hippocampal Long-Term Depression
Physiological tau/tau phosphorylation and LTD
Tau exists mainly in axons,but an emerging body of evidence has now confirmed that a small amount of tau is also present in the somatodendritic compartment under physiological conditions.Dendritic endogenous tau regulates the postsynaptic level of Fyn through its N-terminal sequence (Ittner et al.,2010).Fyn is known to phosphorylate GluN2 subunits of NMDAR,in particular,GluN2B on Tyr1472,to promote the interaction of NMDAR with postsynaptic density protein 95 (PSD-95),thereby resulting in enhanced NMDAR activity (Trepanier et al.,2012).The levels of Fyn in the dendritic spine were abnormally reduced in tau knockout (MAPT–/–) mice (Ittner et al.,2010).The reduction of Fyn leads to a decrease in functional extrasynaptic NMDAR in the hippocampus (Pallas-Bazarra et al.,2019).Conversely,activation of NMDAR causes reversible enhancement of p-tau at specific sites detected by antibodies PHF-1 (Ser396/Ser404),AT180 (Thr231/Ser235),and AT8 (Ser199/Ser202) (Mondragon-Rodriguez et al.,2012).
The relationship between normal tau and synaptic plasticity has only received attention in recent years.Interestingly,endogenous tau and tau phosphorylation at residue Ser396 were discovered to be required for hippocampal LTD but not LTP (Kimura et al.,2014;Regan et al.,2015).Conflicting findings have been reported by Ahmed et al.(2014) that hippocampal LTP rather than LTD was impaired in tau knockout mice under different experimental conditions (Prikas et al.,2022).The most commonly studied form of LTD is induced by Ca2+influx through synaptic and extrasynaptic NMDAR (Papouin et al.,2012;Liu et al.,2013).As mentioned earlier,tau in dendritic spines regulates the dynamic stability of NMDAR via Fyn (Pallas-Bazarra et al.,2019).In tau knockout mice,the level of Fyn in the dendritic spines is reduced,which leads to functional inhibition of extrasynaptic NMDAR (Ittner et al.,2010) and may mediate impaired induction of hippocampal LTD (Kimura et al.,2014).
Regan et al.(2015) discovered that LTD was impaired in cultured hippocampal slice neurons transfected with Ser396A mutant tau,but LTD was preserved in neighboring untransfected neurons.The endocytosis of AMPARs is thought to be important in the expression of LTD triggered by NMDAR activation (Beattie et al.,2000).Regan et al.(2021) further investigated the interaction between tau and protein kinase C and casein kinase II substrate in neurons protein 1(PACSIN1),an F-BAR and SH3-domain-containing protein with established roles in AMPAR endocytosis (Widagdo et al.,2016).Their findings indicate that tau phosphorylation at Ser396/404 decreases the interaction between tau and PACSIN1,which leads to reduced synaptic AMPAR function and structural synapse weakening (Regan et al.,2021).Although the underlying mechanisms are still poorly understood,recentin vivoevidence indicates the important role of LTD in learning and memory (Ashby et al.,2021).Therefore,dendritic tau and reversible phosphorylation of tau may serve pivotal physiological roles in LTD and related cognitive functions (Figure 2).
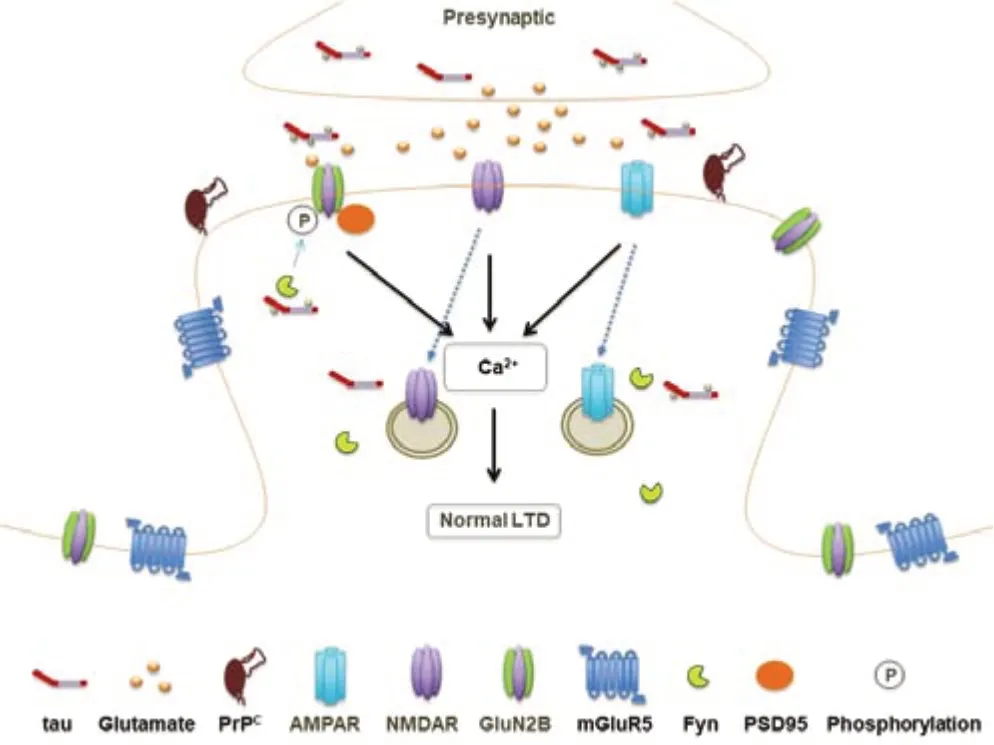
Figure 2|Schematic representation of the putative physiological roles of tau/phosphorylated tau in hippocampal LTD.
Hyperphosphorylated/oligomeric tau and LTD
Neurofibrillary pathology of abnormally hyperphosphorylated tau is a hallmark of AD.Significantly,overexpression of tau constructs carrying phosphomimetic mutations,but not phospho-blocking mutations,facilitated LTD by a weak induction protocol (Mondragon-Rodriguez et al.,2012).The Thy-Tau22 mouse model combines G272V and P301S mutations on the 4-repeat isoform of human tau under the control of the Thy1.2 promoter(Degiorgis et al.,2020).This mouse model shows some key AD-related features such as memory deficit,neuronal death,and neuronal tau pathology including tau hyperphosphorylation beginning in the hippocampus,and the formation of neurofibrillary tangle-like inclusions.Interestingly,only the maintenance of hippocampal LTD was selectively impaired,without affecting LTD induction or LTP (Schindowski et al.,2006;Van der Jeugd et al.,2011;Ahmed et al.,2015).In contrast,insulin-induced hippocampal LTD is facilitated in this transgenic model of tau (Leboucher et al.,2019).On the other hand,Scullion et al.(2019) reported impairment of muscarinic receptor-dependent LTD in the perirhinal cortex of the rTg4510 mice,which overexpress the P301L mutant form of tau.
Growing data suggest that water-soluble aggregates of tau,specifically oligomers,are neurotoxic (Lasagna-Reeves et al.,2012).Interestingly,hyperphosphorylated tau self-assembles form unstructured and amorphous oligomers that can trigger toxicity-promoting pathways (Meng et al.,2022).
Little is known about the properties of tau oligomers and the mechanism by which they may cause damage in tauopathies.As recently reviewed(Niewiadomska et al.,2021),tau oligomers are strongly implicated in mediating neurodegeneration.When applied extracellularly tau oligomers are toxic to neuronal cell cultures and associated with tau uptake into the cell and increased intracellular calcium levels (Pampuscenko et al.,2021).One putative mechanism implicates muscarinic (M1 and M3) subunits of acetylcholine receptors as the initial sites of interaction of tau oligomers with the plasma membrane and in their subsequent clathrin-mediated internalization (Gomez-Ramos et al.,2009;Morozova et al.,2019).In rodents,the injection of tau oligomers induces mitochondrial abnormalities,synaptic dysfunction and,crucially,impairs memory (Lasagna-Reeves et al.,2011;Ondrejcak et al.,2018,2019;Acquarone et al.,2019).
Intriguingly,there is growing evidence that the behavioral toxicity of tau oligomers can be reversible.Doxycycline-induced expression of a tau fragment (∆tau151–421) in P301SxTAU62 mice promotes the formation of hyperphosphorylated and oligomeric tau species,in the absence of fibrillar tau forms or tau tangles,and causes severe motor paralysis after as early as 3 weeks (Ozcelik,2016).Recently,motor paralysis was fully reversed after expression of the tau fragment was stopped,and hyperphosphorylated and oligomeric tau species were no longer detectable (Martinisi,2021).These observations suggest that early targeting of non-fibrillar toxic tau species may represent a therapeutically effective intervention in tauopathies.
Electrophysiological measurements helped uncover several pathophysiological mechanisms mediated by tau oligomers,including altering the intrinsic excitability of pyramidal neurons and modulating both short-and longterm plasticity (Puzzo et al.,2017;Ondrejcak et al.,2018).Certain soluble aggregates of exogenously applied tau,but not monomers or fibrils,selectively disrupt synaptic plasticity in the rat hippocampus (Fa et al.,2016;Ondrejcak et al.,2018,2019).Pre-or post-synaptic intracellular injection of tau oligomers,but not monomers,differentially induce significant changes in action potential dynamics,disruption of basal synaptic transmission,or inhibition of LTP (Hill et al.,2019).Our recentin vivostudy (Ondrejcak et al.,2019) in rat hippocampus showed that soluble Aβ and tau had antagonistic effects on LTD,with Aβ oligomers lowering the threshold for LTD induction and soluble tau aggregates increasing the threshold for LTD induction,but also blocking Aβ-mediated effects.We also reported synergistic effects on LTP,as subthreshold doses of soluble tau aggregates dramatically enhance Aβ oligomer-mediated inhibition of LTP (Ondrejcak et al.,2018).These findings are supported by evidence of both synergistic (DeVos et al.,2018;Busche et al.,2019) and antagonistic (Angulo et al.,2017;Busche et al.,2019) actions of tau and Aβ on network excitability.
Cellular prion protein (PrPC),a 33–35-kDa glycoprotein highly expressed in the brain,has been found to mediate the synaptotoxicity of soluble tau species from AD brain extracts (Ondrejcak et al.,2018),an effect mimicked by extracellular tau species in the secretomes of human induced pluripotent stem cell-derived neuronal trisomy of chromosome 21 model of earlyonset AD (Hu et al.,2018).Importantly,soluble aggregates of full-length recombinant tau share this mechanism of disrupting synapses (Ondrejcak et al.,2019;Corbett et al.,2020).PrPCis located on the outer leaf of the plasma membrane and does not have a transmembrane domain.It interacts with other signaling molecules on the cell membrane,such as metabotropic glutamate receptor subtype 5 (mGluR5) to achieve transmembrane signaling(Haas et al.,2014).Disrupted hippocampal mGluR-dependent LTD has been associated with spatial learning and memory deficits in the Morris water maze in aged Fischer 344 rats (Foster and Kumar,2007) and an Aβ-injected rat model (Hu et al.,2022).
It is not known whether the shared ability to disrupt synaptic plasticity by different preparations and sources of tau is mediated by the same soluble tau species.Soluble aggregates of recombinant tau,used in several studies including ours,were prepared by sonication and appeared to be relatively large assemblies (~50 nm) whereas tau oligomers prepared from crossseeding (Lasagna-Reeves et al.,2010) or by peptide bond formation (Fá et al.,2016) were reported to be smaller (6–20 nm).Somewhat similarly,AD brainderived tau,including tau oligomers,can potently inhibit LTP (Lasagna-Reeves et al.,2012;Fá et al.,2016;Hu et al.,2018) and facilitate LTD (unpublished observations).Aqueous brain extracts are thought to contain a rich range of potentially toxic tau species derived from both intracellular and extracellular sources (Sato et al.,2018).
Overall,accumulating evidence supports the view that the synaptotoxicity of tau is highly aggregation-dependent,with soluble aggregates being very disruptive whereas monomers and insoluble fibrils appear to be relatively inert.Taken together,these studies suggest certain tau species,especially if misfolded and aggregated into oligomers,can cause major pre-and post-synaptic dysfunction and thereby contribute to disease pathogenesis(Figure 3).
Hippocampal Long-Term Depression Promotes Tau Phosphorylation
In a series of elegantly conducted studies,Regan et al.(2015) reported that p-tau396 is required for hippocampal LTD induction.Interestingly,tau phosphorylation at Ser404,but not at residues Ser202/Thr205,was also triggered by a single train of 900 pulses at 1 Hz in acute hippocampal slices from young healthy rats (Figure 4A).
Accumulating evidence suggests that Aβ promotes the generation and spread of tau pathology,in particular tau phosphorylation,but the cellular mechanisms are poorly understood (Busche and Hyman,2020;Scheltens et al.,2021).Previously,we and others reported that Aβ oligomers facilitate a novel form of LTD in the hippocampus (Kim et al.,2001;Li et al.,2009;Hu et al.,2014,2022;O’Riordan et al.,2018b).Taylor et al.(2021) very recently presented evidence that Aβ oligomer-mediated enhancement of the vesicular release of glutamate leads to an increase in the magnitude of LTD induced by LFS in acute hippocampal slices.They further observed,in organotypic hippocampal slices,that either exposure to Aβ oligomers or chronically repeated application of LTD-inducing LFS (every 2 hours per day over 7 days) triggers tau phosphorylation at residues Ser202 and Thr205(p-tau202/205).This study provides valuable,although indirect,mechanistic evidence of synergism between Aβ and tau,the two hallmark proteins in AD (Busche and Hyman,2020).Since the aberrant elevation of p-tau181,p-tau217,and p-tau231 in CSF and plasma were recently proposed to be particularly sensitive markers of preclinical AD,our group investigated the effects of LTD induction on tau phosphorylation at these and other residues in the hippocampus of live rats.We found that LTD-inducing LFS preferentially enhances p-tau181 and p-tau217 in aged,but not young adult,animals.Remarkably,in young adults the same LFS conditioning protocol,when applied soon after intracerebroventricular injection of Aβ,preferentially triggers the enhancement of p-tau181.These new findings indicate that Aβ can shift the age-dependence of tau phosphorylation by LTD-inducing LFS (unpublished data).The enhancement of tau phosphorylation at these residues by LFS appeared more sensitive compared with residues Thr231,Ser202/Thr205,and Ser396 (Zhang et al.,2022b).Of note,a very recent community-based population study indicates that the plasma concentrations of both p-tau181 and p-tau217 increase with age starting between the ages of 65 and 70 years in Aβ-positive participants (Mielke et al.,2022).
The role of glutamate receptors in LTD-associated tau phosphorylation
LTD can be induced by prolonged periods of conditioning stimulation that activate NMDAR and/or mGluR (Collingridge et al.,2010).Regan et al.(2015)discovered that enhancement of p-tau396 by LFS can be blocked by the NMDAR antagonist AP5 in acute hippocampal slices from young healthy rats.When Taylor et al.(2021) chronically stimulated organotypic hippocampal slices either with an NMDAR-dependent protocol or mGluR-dependent protocol only the NMDAR-dependent protocol triggered significant tau hyperphosphorylation at sites detected by the antibody AT8.Whereas the induction ofin vivoLTD at CA3-CA1 synapses is only blocked by combined pretreatment with standard doses of CPP and MTEP (O’Riordan et al.,2018a),injecting either agent alone prevented LFS-triggered increases in p-tau181 and p-tau217 in live aged rats (Zhang et al.,2022b).Apparent discrepancies between these and related findings are likely due to different experimental conditions including live-aged ratsversusacute hippocampal slices or organotypic hippocampal slices from young healthy rats.
mGluR5 is predominantly expressed perisynaptically and extrasynaptically on postsynaptic spines of pyramidal cells in the hippocampal CA1 area.Coactivation of mGluR5 with GluN2B-containing NMDAR located in this vicinity is known to strongly enhance the function of the extrasynaptic NMDAR(Kotecha et al.,2003;Sarantis et al.,2015).Extrasynaptic NMDAR have been considered important mediators of neurotoxicity,while synaptic NMDAR are mainly involved in normal cognitive function (Vieira et al.,2020).Indeed,Sun et al.(2016) found that activation of extrasynaptic rather than synaptic NMDAR on cultured rat cortical neurons for 12 to 24 hours significantly increased tau phosphorylation at residues Ser396 and Ser262.These findings are consistent with a significant involvement of extrasynaptic NMDAR activation in tau pathology.As mentioned earlier,the formation of the Aβ-PrPC-mGluR5 complex has been found to mediate the neuronal toxicity of Aβ oligomers.We reported that Aβ oligomers facilitate mGluR5-dependent LTD in the hippocampusin vivo(Hu et al.,2014,2022;O’Riordan et al.,2018b).Therefore,future studies should examine the role of mGluR and both synaptic and extrasynaptic NMDAR,in tau phosphorylation triggered by LTD-inducing LFS in the presence of synaptotoxic Aβin vivo.
The role of protein kinases/phosphatases in LTD-associated tau phosphorylation
The longest form of human tau contains 85 potential phosphorylation residues.It can be phosphorylated at many different sites by different protein kinases and dephosphorylated by a group of protein phosphatases (Drepper et al.,2020).
Among these protein kinases,glycogen synthase kinase 3β (GSK3β) is widely considered the most important protein kinase in tau phosphorylation.Active GSK3β phosphorylates tau at Ser396,Ser404,Ser199/202,and Thr231 sites (Song and Yang,1995;Rankin et al.,2007).The activity of GSK3β is significantly enhanced in hippocampal LTD (Peineau et al.,2007).Indeed,the GSK3β selective inhibitor CT-99021 abolished the increase of p-tau396 triggered by LTD-inducing LFS in acute hippocampal slices (Kimura et al.,2014).The expression levels of GSK3β have been found to increase with age and in AD pathology (Lauretti et al.,2020;Zhang et al.,2020).GSK3β is the predominant isoform expressed in both human and rodent brains and the function of GSK3α has been much less investigated.Nevertheless,very recent studies indicate the involvement of GSK3α in both NMDAR-dependent and mGluR5-dependent LTD (McCamphill et al.,2020;Draffin et al.,2021).
Cyclin-dependent kinase-5 (Cdk5),a small protein serine/threonine kinase with close structural homology to the mitotic CDKs,has been reported as another important protein kinase for tau hyperphosphorylation (Shah and Lahiri,2017;Cortes et al.,2019;Zhou et al.,2020).Cdk5 directly controls the degradation of NMDAR.Cdk5 phosphorylates Ser1116 of GluN2B,which then leads to suppressed synaptic plasticity and memory (Plattner et al.,2014),while downregulation of Cdk5 improves synaptic plasticity and learning and memory (Plattner et al.,2014;Posada-Duque et al.,2017).Cdk5 activation appears to be required for NMDAR-dependent LTD at CA3-CA1 synapses(Mishiba et al.,2014).
Mitogen-activated protein kinases (MAPK,in particular,p38 MAPK),protein kinase A (PKA),and Ca2+/calmodulin-dependent protein kinase II (CaMKII)are also involved in tau hyperphosphorylation (Lauretti and Pratico,2020;Stefanoska et al.,2022).The MAPK pathway plays a key role in hippocampal synaptic plasticity,in which the activation of p38MAPK is not only involved in NMDAR-dependent (Zhu et al.,2002),but also in mGluR-dependent(Moult et al.,2008) hippocampal LTD.Recent evidence indicates that the activation of p38>Moultin astrocytes is involved in LTD at the hippocampal CA3-CA1 pathway (Navarrete et al.,2019).Sanderson et al.(2016) reported the involvement of protein kinase A,when anchored to the scaffold protein AKAP150,in NMDAR-dependent LTD at CA3-CA1 synapses.While activation of CaMKII was initially considered to participate in hippocampal LTP but not LTD,recent evidence indicates that CaMKII participates in hippocampal LTP and LTD in different activation forms (Coultrap et al.,2014;Szabo et al.,2016).When activated by low-frequency stimulation for LTD induction,CaMKII phosphorylates the Ser567 residues of GluR1,thereby reducing the expression of AMPARs in the postsynaptic membrane.In contrast,when activated by high-frequency stimulation used for LTP induction,CaMKII phosphorylates Ser831 residue of GluR1 and enhances the expression of AMPARs in the postsynaptic membrane (Coultrap et al.,2014).
Protein phosphatase 2A is the major enzyme for the dephosphorylation of tau.It also dephosphorylates Ser845 on AMPAR subunit GluR1 and thereby participates in the induction and expression of hippocampal LTD (Mulkey et al.,1993).Moreover,in protein phosphatase 2A conditional knockout mice,the induction of hippocampal LTD was prevented (Wang et al.,2019).
Aβ oligomers have been shown to increase glutamate release probability(Taylor et al.,2021) and impair the uptake and clearance of synaptically released glutamate (Matos et al.,2008;Li et al.,2009).In addition to increasing the phosphorylation of tau via multiple kinases and phosphatases,the resultant increase in extracellular glutamate levels is believed to cause the Aβ-induced enhancement of LTD (Li et al.,2009;Li and Selkoe,2020;Taylor et al.,2021).The aged hippocampus is especially susceptible to AD pathology(Hou et al.,2019) through numerous putative mechanisms.Amongst these age-related processes,a reduction in glutamate uptake at hippocampal CA1 synapses has been associated with an increase in the activation of both extrasynaptic NMDAR and mGluR,both of which promote LTD (Potier et al.,2010).We propose that age-and Aβ-related pathological facilitation of LTD involves the activation of LTD-associated kinases,such as GSK3α/β,Cdk5,MAPK,protein kinase A,and CaMKII.Increased kinase activation,combined with a decrease in the activation of phosphatases such as protein phosphatase 2A,leads to hyperphosphorylation of tau residues including those not usually phosphorylated under physiological conditions (Figure4B).Hyperphosphorylated tau irreversibly self-assembles into amorphous aggregates and eventually forms neurofibrillary tangles.
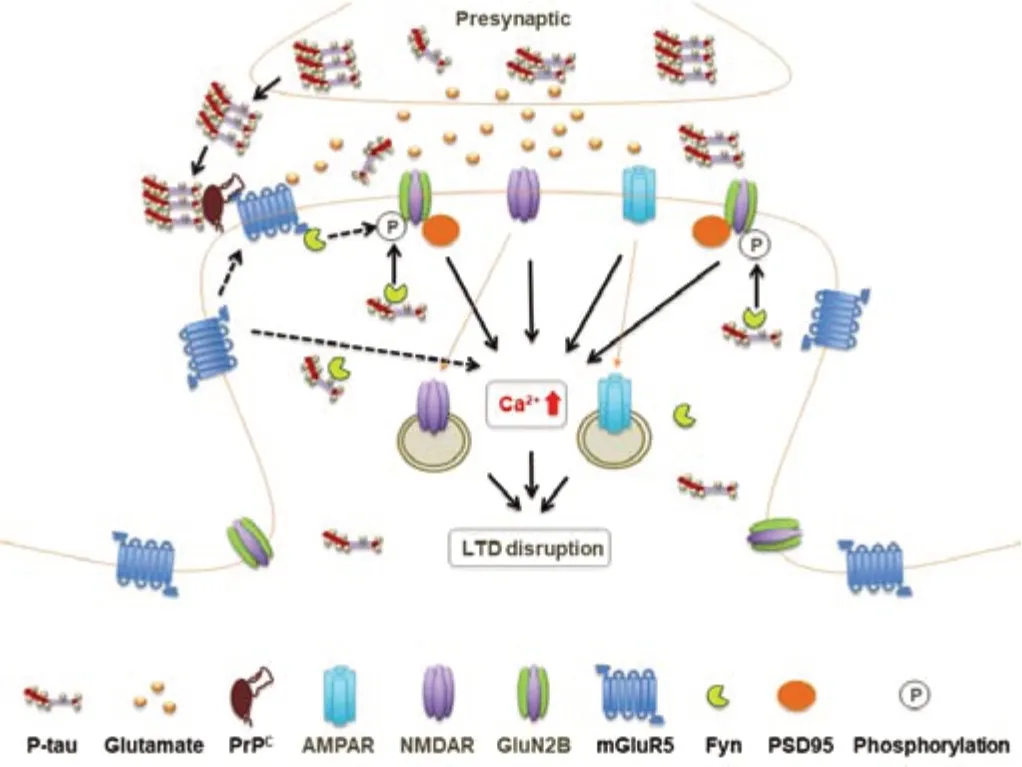
Figure 3|Schematic representation of the putative pathological roles of hyperphosphorylated/oligomeric tau in hippocampal LTD.
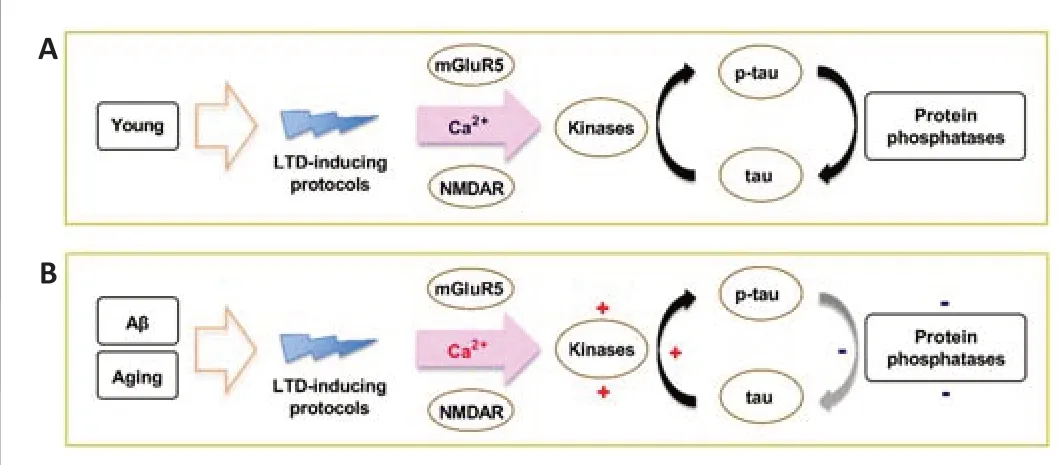
Figure 4|Proposed mechanisms of how LTD-inducing protocols may promote tau phosphorylation under physiological and AD-promoting conditions.
Conclusion and Future Research
There has been increasing research interest in the involvement of tau in AD.A breakthrough has been the discovery that certain plasma p-tau species are particularly sensitive markers of early AD,long before the diagnosis of clinical dementia.Several completed/ongoing clinical trials are targeting tau phosphorylation,but the overall beneficial effects on cognitive symptoms are mixed with reports of common mild to serious side effects.Since endogenous tau and tau phosphorylation may serve pivotal physiological roles,including synaptic plasticity,the patterns of physiological and pathological tau should be carefully distinguished before embarking on the costly development of treatments targeting individual forms of abnormal tau.
Although synaptic plasticity is induced by artificial conditioning protocols in most research,there is extensive and growing evidence that experimentally induced forms of hippocampal LTP and LTD mimic at least some of the core features of naturally occurring forms.Experimentally induced LTP and LTD thus provide a practical means of exploring AD-associated mechanisms of disruption of physiological processes essential for long-lasting changes in circuit connectivity.Such changes include engram strengthening and weakening during memory encoding and maintenance (Whitlock et al.,2006;Magee and Grienberger,2020).Moreover,LTP/LTD-like plasticity can be induced in intact human brain cortex by non-invasive brain stimulation techniques (Mansvelder et al.,2019;Lengali et al.,2021) and as noted in the Introduction,by naturalistic stimulation in both humans and animals.Nevertheless,it should always be borne in mind that artificial stimulation protocols used to induce synaptic plasticity only model aspects of naturally occurring synaptic plasticity.
Whereas the cellular mechanisms of hippocampal LTD have been well documented,the role of LTD in hippocampus-associated learning and memory is still relatively unexplored.In this review,we highlight the unique interaction of tau and LTD based on the recent evidence from our group and others.LTD most likely triggers reversible phosphorylation of tau under physiological conditions,which may play an important role in learning and memory.However,LTD induction protocols can trigger hyperphosphorylation of tau under conditions known to promote AD-related pathology,such as brain aging and the insidious accumulation of toxic forms of Aβ.Given that facilitation of a novel form of LTD has been linked to cognitive decline in our delayed animal model utilizing a single intracerebroventricular injection of synthetic Aβ1–42,tau-LTD interactions in the hippocampus are likely to play a significant role in the progression of AD.For this role to be fully assessed,it will be necessary to validate these mechanistic insights and extend research into related behavioral assessments inin vivomodels of AD.
AD is a complex disease.Tau pathology can be regulated by numerous other factors,often in an Aβ-independent manner.How regulators of AD such as apolipoprotein E,the endocytic system,cholesterol metabolism,and microglial activation influence tau-LTD interactions also needs to be elucidated.Such future research will aid the ongoing evaluation of the core hypothesis of this review that such interactions do indeed play a pivotal role in the progression of AD.
Acknowledgments:We thank Professor Tim Lynch (Mater Misericordiae University Hospital,University College Dublin) for his advice.
Author contributions:NWH and MJR designed and reviewed the manuscript.ZH,TO,PY,and YZ reviewed the literature and wrote the first manuscript.All authors reviewed the manuscript and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons Attribution Non Commercial-Share Alike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Neuro faces of beneficial T cells: essential in brain,impaired in aging and neurological diseases,and activated functionally by neurotransmitters and neuropeptides
- Profiling neuroprotective potential of trehalose in animal models of neurodegenerative diseases:a systematic review
- Cdk5 and aberrant cell cycle activation at the core of neurodegeneration
- Recent advancements in noninvasive brain modulation for individuals with autism spectrum disorder
- Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration
- Cell-based therapeutic strategies for treatment of spinocerebellar ataxias: an update