
废均相催化剂氧化‒络合浸出铑工艺及其动力学
2023-01-21 00:57丁云集李佳怡郑环东崔言杰刘波张深根
工程科学学报 2023年2期
丁云集,李佳怡,郑环东,崔言杰,刘波,张深根✉
1) 北京科技大学新材料技术研究院金属材料循环利用研究中心,北京 100083 2) 北京科技大学顺德创新学院,佛山 528399 3) 有研资源环境技术研究院(北京)有限公司生物冶金国家工程实验室,北京 101407
铑具有高催化活性、高稳定性和抗腐蚀性等独特的物化性能,广泛应用于催化领域[1–2],如石油化工[3]、汽车尾气净化[4–5]、羰基合成均相[6–7]等.羰基合成均相催化剂(又称威尔金森催化剂)主要包括三苯基膦氯化铑、三苯基膦羰基乙酰丙酮铑两种,是活性最高的均相加氢催化剂[8],广泛应用于不饱和烃和不饱和聚合物的C=C 选择性催化加氢[9–10].含铑均相催化剂因杂质的积累、羰基铑簇及芳基铑的形成致失活报废,年报废量达到1000 t以上,是铑的重要来源.含Rh 废均相催化剂由于含有大量有机物,有刺激性气味,存在健康和环境风险[11–12],属于HW50 类危险废物,如何实现铑的资源化利用利用成为人们研究的重点.
均相催化剂的工业应用比多相催化剂晚,其再生研究也相对较晚.目前,从废均相催化剂中回收铑的方法主要有火法和湿法工艺.火法是将废均相催化剂和碱金属或碱土金属化合物(如碳酸钠)混合,在高温下进行焚烧,使Rh 富集在焚烧渣中,然后通过活化溶解、分离提纯处理铑灰,最终还原获得金属铑[13].如蒋凌云等[14]利用“蒸馏–焙烧”混合工艺处理报废均相催化剂含Rh 灰分,再对其进行无机酸消解回收铑;杜继山[15]将含铑废有机液体进行自然焚烧后与铝粉混合焙烧,然后盐酸除铝,灰渣用王水溶解,再提纯成RhCl3.该工艺流程短、有机物杂质去除效果好,但焚烧过程Rh 随烟气挥发致回收率低,且三苯基膦燃烧形成剧毒的氧化磷烟气,危害人体生命健康[16].
为降低污染,提高Rh 的回收率,Yang等[17]采用无机酸(硫酸、硝酸、高氯酸、盐酸)和氧化剂的混合溶液消解废铑均相催化剂,然后碱中和消解溶液,盐酸溶解,Rh 的回收率可达97%.蒋凌云等[18]加入浓硫酸将废铑均相催化剂炭化,然后加入碱金属硝酸盐消解直至体系溶液清澈透明,有机物以CO2形式除去,Rh 以可溶性盐的形式存在于溶液中.无机酸消解法回收率高,但酸耗高、反应剧烈、有NOx溢出,二次污染严重.Davidson 和Fieselmann[19]利用O2氧化废铑均相催化剂,将Rh 转入无机相中,但该方法浸出率不稳定而且O2流速无法精确控制,但该结果表明可通过氧化将废均相催化剂中的Rh+氧化成Rh3+,减少有机配体对Rh 的束缚,从而将Rh 从有机相中解离.
基于上述研究背景,本文提出了采用H2O2作为氧化剂,用于氧化铑膦络合物,破碎Rh–P 化学键,从而实现Rh 的绿色解离;采用Cl–络合解离后的Rh3+形成RhCl63–,降低体系吉布斯自由能,提高Rh 的解离效率;同时为降低体系酸浓度,减少甚至避免生成Cl2,添加NaCl 提高体系Cl–浓度.为降低试剂的消耗,本文首先通过蒸馏将低熔点有机物回收,然后采用HCl–NaCl–H2O2低酸体系氧化–络合浸出Rh,研究了蒸馏温度、Cl–浓度、H2O2用量、H+浓度、反应时间对Rh 浸出率的影响规律,通过响应面法优化了上述工艺参数,最后通过分光光度法研究了Rh 的浸出动力学,计算了其活化能,为废均相催化剂中Rh 的高效浸出提供了理论基础和技术支撑.
1 实验部分
1.1 实验原料
本实验所用含铑废均相催化剂来自云南某贵金属回收企业,通过电感耦合等离子体发射光谱仪(ICP-OES)测定其Rh 质量浓度为564.36 mg∙L–1.H2O2为氧化剂、盐酸和NaCl 为络合剂,NaOH 用于沉淀Rh,上述试剂均为分析纯.
1.2 实验方法
废铑均相催化剂的回收主要包括蒸馏、氧化络合、沉淀三个步骤,如图1 所示.
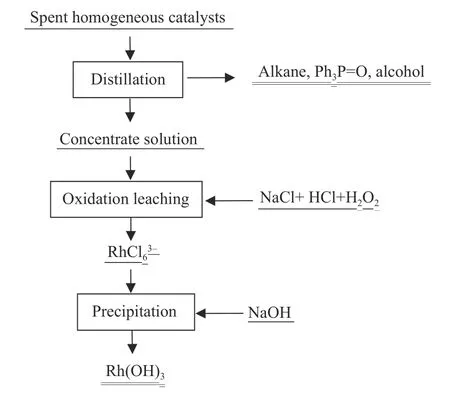
图1 废铑均相催化剂的回收工艺流程图Fig.1 Schematic of the recovery process of spent Rh homogeneous catalysts
(1) 蒸馏:首先将20 mL 废铑均相催化剂置于可恒温控制的电加热套中蒸馏,直到没有明显的冷凝物产生,蒸馏温度240~320 ℃,蒸馏产物经冷凝回收,蒸馏浓缩液用于提取Rh,蒸馏过程示意图如图2 所示.
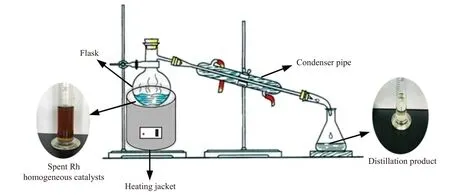
图2 蒸馏过程示意图Fig.2 Diagram of the distillation process
(2)HCl–NaCl–H2O2体系氧化络合浸出Rh:浸出实验均在250mL三口烧瓶中进行,将反应器置于水浴中,搅拌速度350r∙min–1,水浴温度90℃.将10mL蒸馏浓缩液以及0~1.0mol∙L–1盐酸、0~5.0mol∙L–1NaCl放入三口烧瓶中,相比固定为1∶1,H2O2用量为蒸馏浓缩液体积分数的10%~45%,反应时间1.0~6.0h,反应结束后,采用分液漏斗分相,分析水相中Rh 的含量,其浸出率计算公式如下:
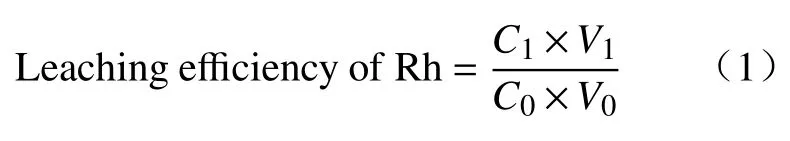
式中:C1为浸出液中Rh 的浓度,V1为浸出液的体积,C0为蒸馏浓缩液中Rh 的浓度,V0为浓缩液体积(10 mL).
(3) 沉淀:采用NaOH 调节水相中pH值,沉淀、过滤得到Rh(OH)3,再经分离提纯得到Rh 产品.
1.3 材料表征
通过傅里叶变换红外光谱(NEXUS670FT-IR,Thermo Nicoiet,America)分析废铑均相催化剂和蒸馏产物中有机物的种类;通过紫外−可见光分光光度计(TU-1900,PERSEE,China)在320~850 nm范围内测定铑氯络合物的最大吸收波长以及在该波长下不同反应时间的吸光度值;采用ICP-OES(Optima 8000,PerkinElmer,America)检测溶液中的RhCl63–含量.
2 结果与讨论
2.1 蒸馏
图3(a)为废均相催化剂的红外光谱分析结果,可以看出,3452.34 cm–1处的吸收峰主要是O–H 的伸缩振动,1186.14 cm–1处的吸收峰主要是C–C、C–O 官能团的伸缩振动引起,表明废均相催化剂中含有部分的游离醇;2966.67、1458.08 和1377.08 cm–1处的强吸收峰对应于烷烃基团(如C–H、C=C,C–C等),738.69 和1726.17 cm–1处的特征带分别由苯环的弯曲振动和P–C 伸缩振动造成的,结合含铑均相催化剂的服役环境,推测废均相催化剂由醇、饱和烷烃、三苯基膦、三苯基氧化膦组成.图3(b)为280 ℃下蒸馏2 h 得到的蒸馏产物的红外光谱分析,可知其主要成分为烷烃、醇.
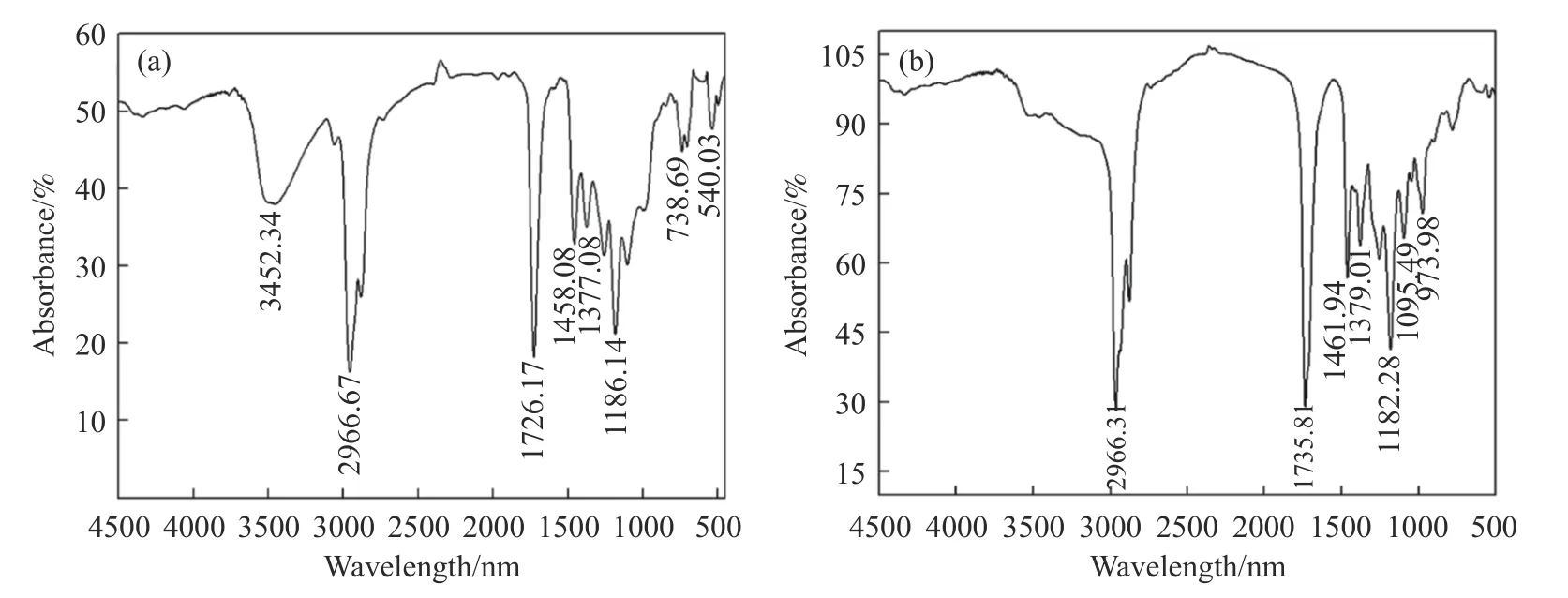
图3 红外分析光谱.(a)废均相催化剂;(b)蒸馏产物Fig.3 Infrared absorption spectrum recorded: (a) spent RCHC;(b) distillate
饱和烷烃和醇的沸点通常低于240 °C,为尽量减少试剂消耗和有机杂质与Rh 的结合,提高Rh的浸出率,取20.0 mL 废均相催化剂,在240~320 °C条件下蒸馏效果4 h,蒸馏效果如图4 所示.随着蒸馏温度的升高,蒸馏产物中Rh 的浓度逐渐增加,由240 ℃的4.15 mg∙L–1增加至320 ℃的53.81 mg∙L–1;同时蒸馏产物体积从5.3 mL 增加至11.5 mL.综合考虑蒸馏产物中Rh 的浓度与蒸馏浓缩效果,确定较适宜的蒸馏温度为260 ℃.
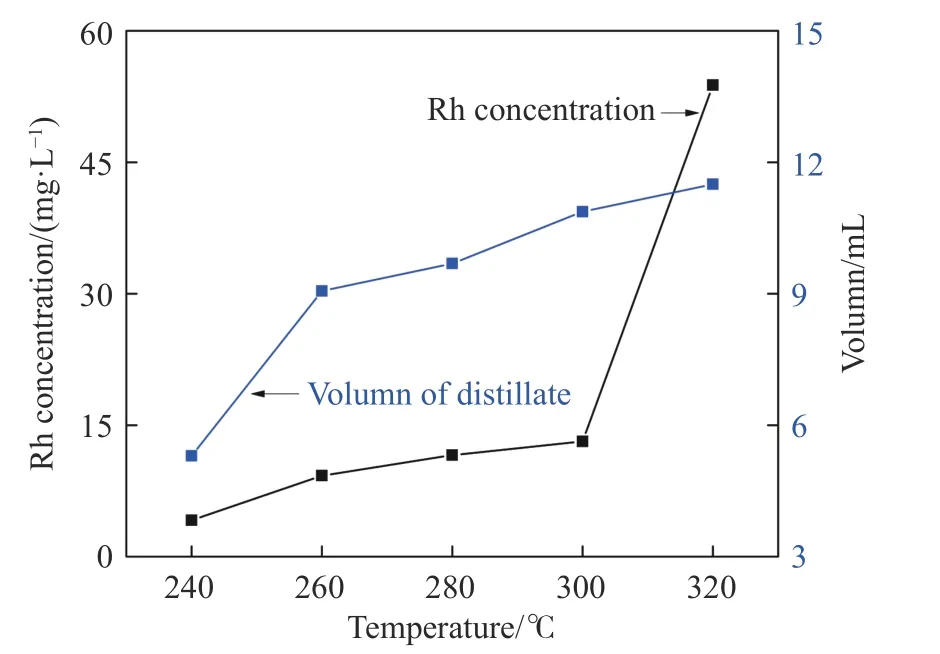
图4 240~320 ℃下蒸馏产物的体积及Rh 浓度Fig.4 Volume of distillate and concentration of Rh at temperatures ranging from 240–320 ℃
2.2 氧化络合浸出Rh
废均相催化剂经260 ℃蒸馏4 h 后得到蒸馏浓缩液,采用HCl–NaCl–H2O2体系氧化络合浸出Rh,讨论了反应时间、H2O2用量、Cl–浓度、H+浓度对Rh 浸出率的影响.
图5(a)为当c(H+)=1.0 mol∙L–1、H2O2用量为蒸馏浓缩液体积分数的20%,在90 ℃下反应4 h 条件下Rh 的浸出率随Cl–浓度变化关系,随Cl–浓度从1.0 mol∙L–1增加到2.0 mol∙L–1,Rh 浸出率从23.0%迅速提高到45.6%;当Cl–浓度从2.0 mol∙L–1增加到5.0 mol∙L–1时,Rh 的浸出率从45.6%缓慢增加到62.9%.这是因为Cl–浓度的提高,有利于Rh3+络合形成水溶性的RhCl63–.而当Cl–浓度大于2.0 mol∙L–1时,浸出效率的提高速度逐渐减慢,这可能是由于在RhCl63–浓度变高后,RhCl63–与体系中残留的三苯基膦(Ph3P)反应回到有机相,化学方程式下:
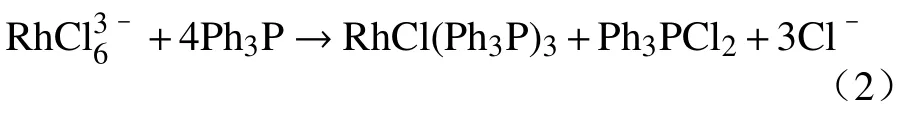
图5(b)为当c(H+)=1.0 mol∙L–1、c(Cl–)=5.0 mol∙L–1,在90 ℃下反应4 h 条件下Rh 浸出率随H2O2用量变化关系.当H2O2加入量为蒸馏浓缩液体积分数的10%时,Rh 浸出率仅为10.8%;随着H2O2用量增加到30%时,Rh 浸出率迅速升高到80.7%,然后趋于平缓.Rh 浸出率在前期明显增加是因为增加H2O2用量可以在低浓度时促进强氧化自由基∙OH的生成[20],从而更容易破坏三苯基膦铑络合物结构,强化Rh–P 键的解离.然而,高浓度的H2O2对∙OH 有猝灭作用,从而生成∙OH2[21],使氧化效率降低,从而使Rh 浸出效率趋于平缓.因此,H2O2的较佳用量为蒸馏浓缩液体积分数的30%.
图5(c)为当c(H+)=1.0 mol∙L–1、c(Cl–)=5.0 mol∙L–1、H2O2用量为蒸馏浓缩液体积分数的30%,在90 ℃下Rh 浸出率随反应时间的关系.在1~4 h 范围内Rh 浸出率与反应时间成正相关,从1 h 的50.07%提高至80.7%,然后随反应时间延长Rh 浸出率降低.这可能是因为4 h 后H2O2已完全反应,体系中残留的三苯基膦与RhCl63–反应生成三三苯基膦氯化铑回到有机相中,从而降低了Rh 的浸出率,原理见式(2).
图5(d)为c(Cl–)=5.0 mol∙L–1、H2O2用量为蒸馏浓缩液体积分数的30%,在90 ℃反应4 h 条件下H+浓度对Rh 浸出率的影响.可知Rh 的浸出率随H+浓度的增加先上升后下降,当H+浓度超过1.0 mol∙L–1后Rh 浸出率急剧下降.这可能是在强酸体系下,Cl–与H2O2发生反应生成Cl2逸出,降低了体系中Cl–浓度,同时导致有效H2O2用量减少,无法氧化废均相催化剂的Rh+致其浸出率低.
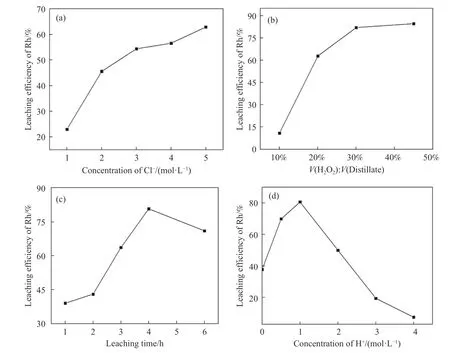
图5 氯离子浓度(a)、双氧水用量(b)、反应时间(c)、H+浓度(d)对Rh 浸出率的影响Fig.5 Effects of Cl– concentration (a),H2O2 dosage (b),leaching time (c),and H+ concentration (d) on Rh leaching efficiency
2.3 响应面法优化实验设计
通过上述单因素实验结果可知,在较优的工艺参数:c(H+)=1.0 mol∙L–1、c(Cl–)=5.0 mol∙L–1、H2O2用量为蒸馏浓缩液体积分数的30%、90 ℃下反应4 h,Rh 的浸出率为80.7%.该浸出率对Rh 的回收并不理想,且单因素实验不能很好体现氧化络合浸出体系中各因素的交叉作用对Rh 浸出率的影响,响应曲面法可在有限次的实验模拟得到优化后的工艺参数,且能反映浸出体系内各影响因素的交叉作用[22–23],得到Rh 浸出率与各影响因素间的关系表达式,实现对Rh 氧化络合浸出的优化.根据单因素实验结果,固定c(H+)=1.0 mol∙L–1、反应温度90 ℃、反应时间4 h,将Cl–浓度、H2O2用量和反应时间分别设定为3.0~7.0 mol∙L–1、20%~40%和2~6 h,建立响应面模型确定上述三个因素间的交互作用.采用CCD 中心组合设计(Central composite design)模型设计3 因素3 水平优化实验,通过Design Expert 8.0 对上述三个因素之间的交叉作用和对Rh 浸出率的影响进行分析,得到下列回收方程:
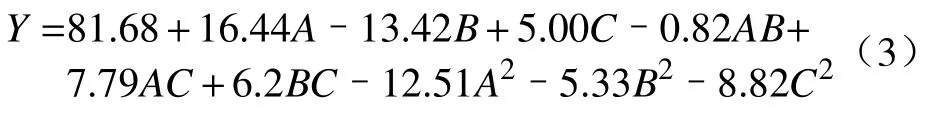
式中:Y为响应值(Rh 浸出率,%),A为H2O2用量,B为Cl–浓度(mol∙L–1),C为反应时间(h).从式(3)可以看出,A和C的线性因子以及BC相互作用对浸出率有促进作用,而AB和AC的相互作用以及B的水平对浸出率有负影响.回归模型的显著性分析结果和相关性分析分别如表1 和表2 所示.
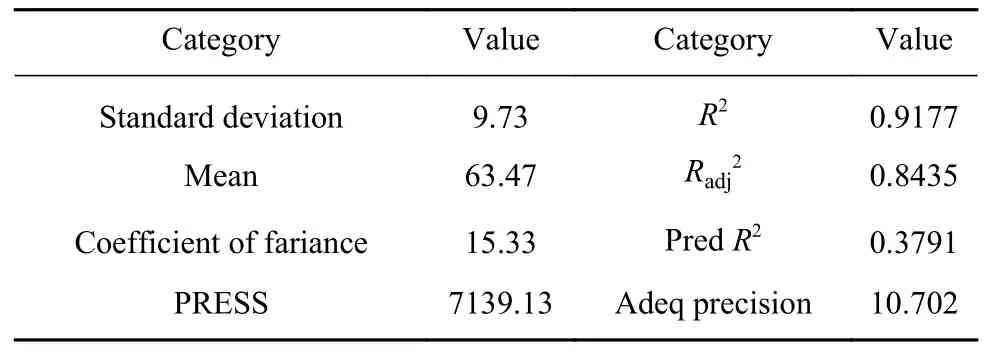
表2 响应曲面模型的相关性分析Table 2 Correlation analysis of response surface method
Design Expert 8.0 软件中方差分析,在ANOVA中F值组间均方与组内均方的比值,p值为相应F值下的概率值,当Prob>F小于0.05 时表明该模型的模拟精准、效果好[24].表1中F值为12.38,说明Rh 氧化浸出模型是显著的,p值为0.0003,表明该模型精准度高;而其复相关决定系数R2=0.9177,表明该模型拟合度良好,失拟项的p值小于0.0001,可能是由于响应值集中导致.调整复相关系数Radj2=0.8435,表明 84.35%的实验数据可以用该模型进行解释.信噪比(Adeq precision)值为有效信号与噪声的比值,当其大于4 时是较为理想的.本模型的信噪比为10.702,表明该模型较为理想.从方差分析可以看出一次项A的 Prob>F值小于0.0001,B的Prob>F值为0.0005,C的Prob>F值 为0.0866,表明A因素影响极其显著,B的影响较为显著,C影响较小;交叉项AB、BC的影响不显著,AC较为显著;二次项中A2、C2影响显著,B2影响不显著.因此,在本研究Rh 的氧化浸出条件下,各因素对Rh 浸出率的影响程度大小为H2O2用量>Cl–浓度>反应时间.
图6 为不同影响因素交互作用下对Rh 的浸出率影响.在曲面图的宽红色区域,表示Rh 的溶解率大于80%,黄色区域表明Rh 浸出率为70%~80%;绿色区域表明Rh 浸出率小于60%.随着A的增加,Rh 浸出率持续升高如图6(a);当B和C固定在3.0 mol∙L–1和4 h时,A从20%提高到40%,Rh浸出率从33.6%提高到86.27%;固定A为20%、C为4 h,随着B的增加,Rh 浸出率增长缓慢如图6(c),而且同时增加A和B的值,Rh 浸出率还有下降的趋势,可能原因是在高温下两者用量增加后,会促进H2O2和Cl–溶液反应生成Cl2逸出,无法充分氧化铑膦中的Rh+,使其仍保留在有机相中;从总体上看浸出时间C对Rh 浸出率影响较小.通过Expert Design 软件分析,系统给出的优化工艺参数:双氧水用量37%、Cl–浓度3.0 mol∙L–1、反应时间4.5 h,Rh 浸出率预测值为96.29%.通过三次平行实验验证,在模拟工艺参数下Rh 浸出率为98.23%,超过预测值.
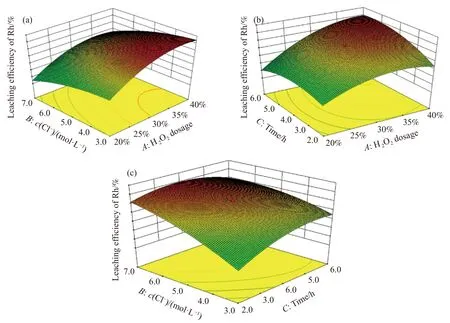
图6 不同因素相互作用对Rh 浸出率影响的三维响应图.(a) C,time=4 h;(b) B, c(Cl–)=5 mol∙L–1;(c) A,H2O2 用量为30%Fig.6 Response surface plots for the interaction effects on the Rh leaching rate: (a) C,time=4 h;(b) (b) B, c(Cl–)=5 mol∙L–1;(c) A,H2O2 dosage of 30%
2.4 Rh 氧化络合浸出动力学
通过上述分析,影响反应速率的因素有反应温度、反应时间、Cl–浓度、双氧水用量等,且随着四种因素的增加,浸出率在前期均有明显的升高.从微观角度分析,上述因素在反应过程中均起着不可替代的作用.由于Rh 的氧化络合浸出反应为液液非均相反应,根据双膜理论,液液非均相反应主要包括无机离子、有机物分子分别通过液膜扩散到相界面的扩散过程及界面化学反应.通过响应面法分析可知,高温(90 ℃)下H2O2用量和Cl–浓度对Rh 浸出率的相关性大于反应时间,表明高温体系反应分子的扩散速率加快,界面化学反应为速度控制步骤.因此,溶液中H2O2氧化浸出废Rh 均相催化剂的反应过程可以分为以下五个步骤(如图7 所示):
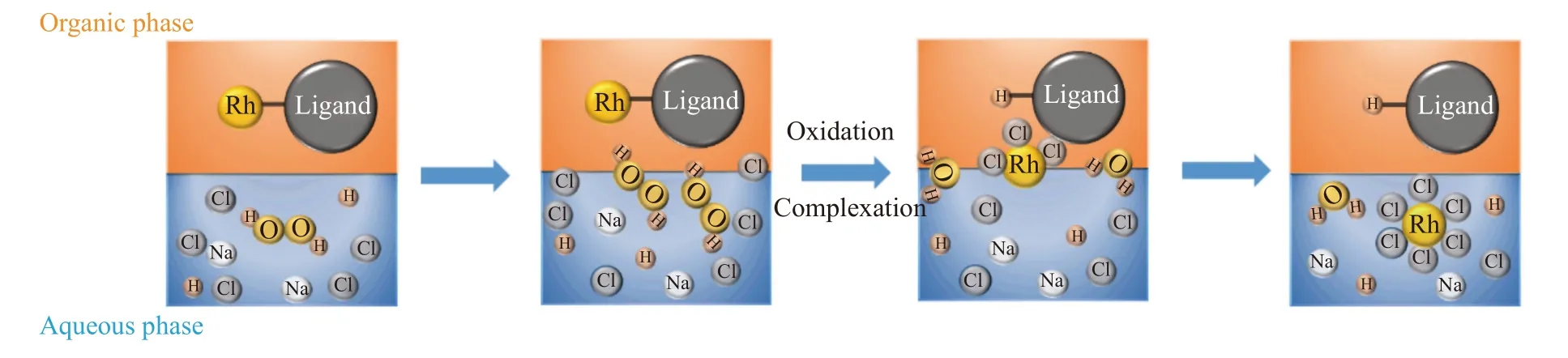
图7 废均相催化剂氧化浸出Rh 的反应示意图Fig.7 Diagram of oxidation leaching of Rh using spent homogeneous catalysts
(1)组元[H2O2+H++Cl–]由水相穿过水相一侧边界层向水相/有机相界面迁移;
(2)组元[Rh–CnHm]由有机相穿过有机相一侧向有机相/水相界面迁移;
(3)在相界面上发生化学反应;
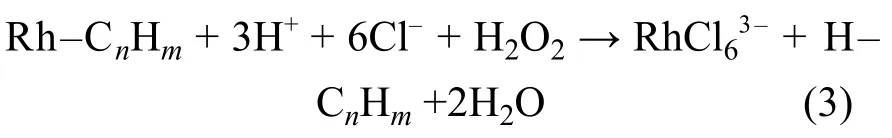
(4)反应产物[RhCl63–+H2O]由有机相/水相界面向水相迁移;
(5)反应产物[H–CnHm]由水相/有机相界面向有机相迁移.
为验证界面化学反应是否为速率控制步骤,使用分光光度法测定Rh 浸出活化能.将不同浓度的铑氯络合物溶液在200~850 nm 光谱下进行扫描(如图8 所示),发现铑氯络合物在498 nm 得到最大吸收峰,然后在498 nm 处测量不同反应时间溶液的吸光度值,确定RhCl63–的浓度.
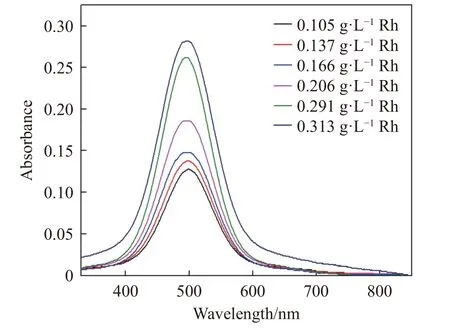
图8 不同浓度的RhCl63–溶液的紫外吸收光谱Fig.8 Ultraviolet absorption spectra of RhCl63− solution at different concentrations
根据不同温度下溶液的吸光度A随时间的变化曲线,整理分析反应动力学规律.在准一级反应条件下(即:[氧化剂]0>>[还原剂]0),根据反应动力学方程式:
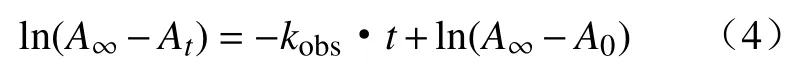
式中:At和A∞表示在时间为t和 ∞时的吸光度,kobs为表观速率常数.取反应过程中的15At~20At,以ln(A∞−At)对时间t作图得到拟合直线(如图9 所示),通过求其斜率即可得出不同温度下的表观速率常数kobs.
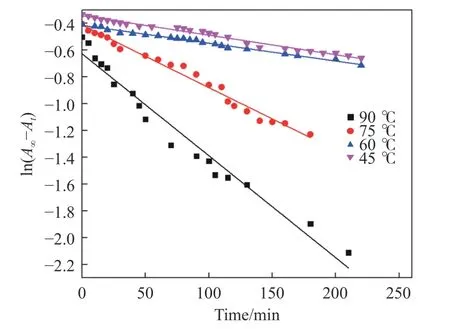
图9 ln(A∞−At)与反应时间的关系Fig.9 Relationship between ln(A∞−At) and reaction time
由上图可知在不同温度下ln(A∞−At)均呈下降趋势,而且温度越高,直线的斜率越大.根据表3,在45°C 和60 °C 下直线的斜率相差不大,四条直线的拟合相关系数均大于0.95,表明ln(A∞−At)与kobs·t线性拟合度良好,Rh 氧化络合浸出符合准一级反应条件.
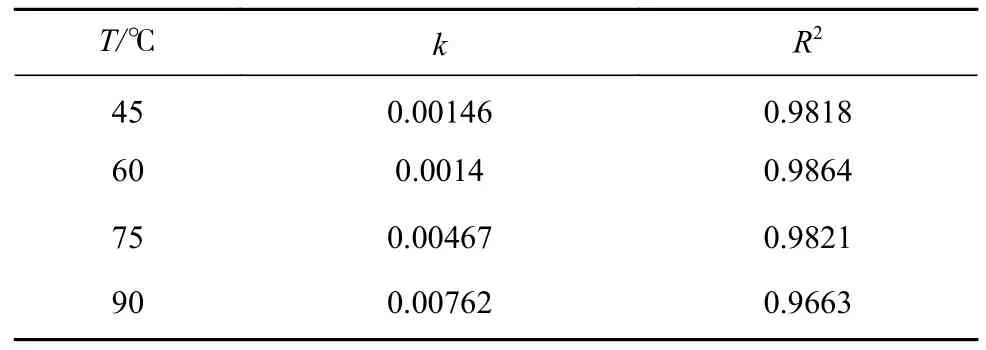
表3 不同温度下Rh 氧化络合浸出动力学参数Table 3 Kinetic parameters of the chemical reaction control model for Rh leaching at different temperatures
表观反应速率常数和温度之间的关系可用Arrhenius 公式表示[25]:
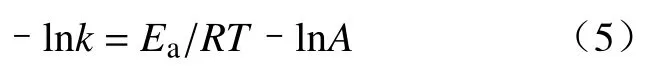
式中:A是指数前因子;Ea为表观活化能,kJ∙mol–1;R为理想气体常数,8.314 J∙mol–1∙K–1;T为绝对温度,K.
将表3 中得到的不同温度下的kobs值代入式(5),得到–lnkobs对1/T的关系,如图10 所示.线性拟合相关系数R2=0.901,根据斜率求得Rh 氧化络合浸出表观活化能为39.24 kJ∙mol–1,为化学反应控速.表明可通过提高H2O2和Cl–浓度降低反应吉布斯自由能,从而强化反应过程,与响应曲面法结果吻合.
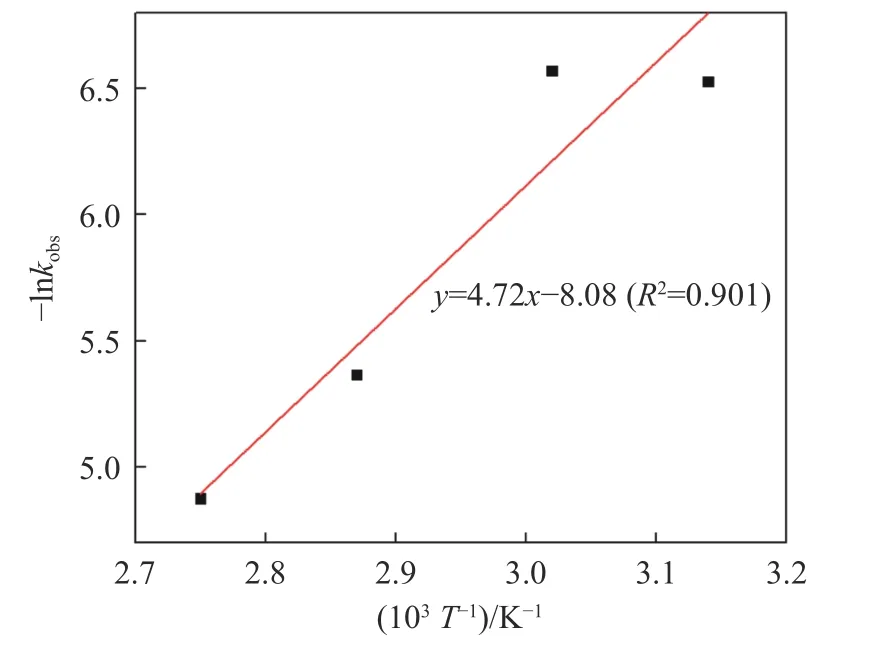
图10 40~90 ℃反应条件下Rh 氧化浸出的Arrhenius图Fig.10 Arrhenius plot for oxidation leaching of Rh at temperatures of 40–90 ℃
3 结论
本文以废均相催化剂为研究对象,考察了蒸馏温度、Cl–浓度、H2O2用量、H+浓度、反应时间等对Rh 的回收率影响,并采用响应曲面法优化了Cl–浓度、H2O2用量和反应时间等工艺参数,揭示了Rh 氧化络合浸出动力学行为,主要结论如下:
(1)采用红外光谱分析确定了废均相催化剂主要由醇、饱和烷烃、三苯基膦、三苯基氧化膦等组成,在260 ℃下蒸馏去除大部分的醇和饱和烷烃,减少后续氧化浸出的试剂消耗和有机物对Rh的束缚,为Rh 的高效回收提供前提.
(2)通过H2O2氧化解离Rh–P 化学键,将Rh+氧化为Rh3+并与Cl–形成RhCl63–进入溶液中,实现了Rh 的氧化络合浸出.采用响应曲面法优化了Rh 浸出工艺,各因素对Rh 浸出率的影响程度为H2O2用量>Cl–浓度>反应时间;优化的浸出条件:双氧水用量为蒸馏浓缩液体积分数的37%、Cl–浓度3.0 mol∙L–1、90 ℃反应4.5 h,Rh 浸出率为98.23%.
(3)采用分光光度法分析了Rh 的浸出动力学行为,表明RhCl63–溶液的最大吸收波长为498 nm,HCl–NaCl–H2O2体系氧化络合浸出Rh 的活化能为39.24 kJ∙mol–1,符合界面化学反应控速模型.
猜你喜欢
中草药(2022年19期)2022-10-14
农业研究与应用(2021年3期)2021-08-23
科学与财富(2021年33期)2021-05-10
首都食品与医药(2020年24期)2020-12-22
环境卫生工程(2020年3期)2020-07-27
天然气与石油(2019年4期)2019-09-10
中国资源综合利用(2019年6期)2019-01-21
中国钼业(2019年2期)2019-01-19
汽车零部件(2018年5期)2018-06-13
安徽农业科学(2017年1期)2017-07-10
