
先天性左侧颈内动脉缺失继发帕金森综合征一例并文献复习
2023-01-23 09:18孙小玲李志军
中国全科医学 2023年3期
孙小玲,李志军
先天性颈内动脉缺失是一种罕见的颈内动脉(ICA)先天性发育异常,发病率不到0.01%[1]。由于在胚胎发育过程中颈动脉血管与ICA关系密切,先天性ICA缺失常与颈动脉闭锁以及ICA相关结构异常有关,如眼动脉起源变异、垂体发育不全、交感神经发育不良、侧支循环通路发育异常等[2]。这种疾病的临床症状谱从无症状到Horner综合征、视野缺失、三叉神经痛、搏动性耳鸣、记忆力障碍/痴呆、短暂性脑缺血发作(TIA)、颅内动脉瘤、蛛网膜下腔出血 (SAH)、垂体功能减退和多器官发育畸形等。先天性ICA缺失在年轻患者中多表现为发育迟缓或SAH,在老年患者中通常表现为TIA[2]。在上述情况中,以颅内动脉瘤的负担最重[3],相关报道也较多。但与先天性ICA缺失相关的帕金森综合征的报道较少。现报道1例先天性ICA缺失伴Horner综合征以及动脉瘤,且继发帕金森综合征的病例,并进行文献复习,以提高临床医生对这一罕见疾病的认识。
1 病例简介
患者,男,68岁,因“右侧肢体乏力3年,加重1个月”于2015-06-03首诊于湖北省直属机关医院。患者于2012年开始逐渐出现右下肢体乏力沉重感,行走动作缓慢;后渐发展至右上肢沉重乏力,间断手抖。无二便障碍。既往有高脂血症、右下肢动脉血栓病史;否认睡眠障碍等病史。入院查体:左侧眼裂小,左瞳孔<右瞳孔,对光反射正常,左侧面颈部干燥无汗。右侧肢体肌力Ⅴ-级,右侧肌张力轻度增高,右下肢腱反射稍增高,右侧Barbinski征阳性。结合患者慢性进行性加重的病程,左侧Horner征阳性,右侧锥体束征和锥外系体征,初步考虑左侧颈部交感神经或脑干/锥体外系病变可能。入院后,患者血、尿、便常规及血糖、电解质、凝血四项、C反应蛋白、D-二聚体、甲状腺功能、叶酸、维生素B12、铜蓝蛋白、重金属、风湿、类风湿、免疫、肿瘤全套等检查结果均正常。颅脑磁共振检查(MR)示脑萎缩,颅内未见异常信号。经颅多普勒检查(TCD)提示左侧ICA闭塞可能。进一步行头颈部CT血管造影(CTA)显示:左侧颈总动脉平颈2椎体处分出左颈外动脉,左ICA缺如,其顶端形态平钝光滑,无鸟嘴样、梭形改变(图1);其远端直至颅内,未见延续血管及血流;左ICA孔内未见血管(图2),提示:(1)左侧ICA先天缺失,(2)左ICA-C7段小动脉瘤以及右锁骨下动脉起始部瘤样扩张。综合患者的症状、体征以及影像学结果,考虑为左侧先天性ICA缺失所致,最后诊断考虑为:(1)左侧先天性ICA缺失;(2)帕金森综合征可能,帕金森病待排;(3)颅内动脉瘤。建议患者予以左旋多巴等对症治疗,患者拒绝后出院。
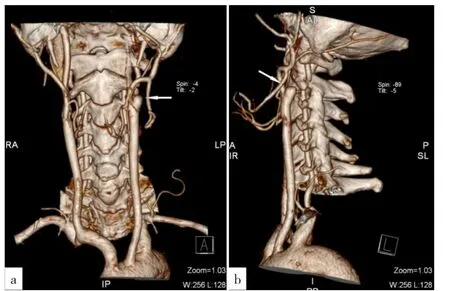
图1 患者头颈部CT血管造影图Figure 1 CT angiography of the patient's head and neck
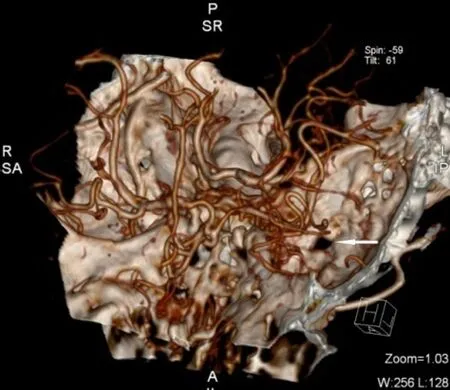
图2 患者颅脑CT血管造影图Figure 2 CT angiography of the patient's brain
后续对该患者进行了7年随访,随访过程中患者出现右侧上、下肢沉重僵硬感以及右手抖动逐渐加重,右手精细动作受累,动作迟缓,姿势步态异常,患者自述“左、右像两个人”,并自感记忆力下降。患者先后加用多巴胺受体激动剂、左旋多巴、安坦、雷沙吉兰等治疗,疗效均欠佳。近期患者再次门诊复诊,查体示:慌张步态,面部表情减少,左侧Horner综合征,右侧肌张力明显增高,肢体协同动作减少,右侧肢体轻度失用。帕金森综合评分量表(UPDRS)评分为52分,H-Y分期为3期。认知功能评分显示,简易精神状态检查量表(MMSE)评分为20分,蒙特利尔认知评估量表(MoCA)评分为20分,主要表现为执行功能下降。复查颅脑磁共振成像检查(MRI)示:脑萎缩(左侧顶叶、额叶萎缩比右侧明显)。复查CTA仍示左侧ICA先天缺如以及左ICA-C7段小动脉瘤以及右锁骨下动脉起始部瘤样扩张。回溯患者近10年的病史,主要特点为:慢性进行性加重的右侧肢体沉重、乏力、僵硬,逐渐出现右侧肢体失用以及认知功能障碍;查体表现为面具脸,慌张步态,右侧锥体外系体征,以及对左旋多巴等对症治疗效果不佳。根据《中国帕金森病的诊断标准(2016版)》[4],该患者在发病10年后仍局限于单侧受累,且对左旋多巴治疗反应欠佳,排除原发性帕金森病,考虑为帕金森综合征。结合患者既往左侧先天性ICA缺如、左侧Horner综合征、颅内动脉瘤等,病因诊断考虑先天性ICA缺失相关的血管性帕金森综合征(vascular Parkinsonism,VaP)。
2 文献复习
计算机检索PubMed以及万方数据知识服务平台,检索时间为建库至2022年4月,以“先天性ICA缺失(congenital absence of ICA)”“帕金森综合征(Parkinsonism)”“血管性帕金森综合征(Vascular Parkinsonism)”为关键词进行检索,未发现相关报道。增加关键词“ICA闭塞(ICA occlusion )”“ICA狭窄(ICA stenosis)”后进一步检索,发现相关报道[5-11],见表1。
共检索到与ICA重度狭窄/闭塞且合并明确的锥外系症状体征的患者10例,加之本例先天性ICA缺失病例报道共11例,其中男8例、女3例;年龄41~81岁,平均年龄(68.2±10.7)岁。大部分患者为慢性起病(8例),2例(例5,例6)锥体外系症状发生在手术后急性偏瘫后。所有病例存在明确的ICA重度狭窄/闭塞/缺失;双侧累及1例,其余均为单侧累及,左侧7例,右侧3例。既往史多合并高血压、糖尿病、高脂血症等,症状特点表现以锥体束、锥体外系(局限性、不对称性)以及皮质认知功能受累(失用、执行功能障碍)为主。2例患者合并共济失调,2例患者合并肌阵挛,1例癫痫可能。颅脑MR多表现为ICA狭窄相应供血区局部脑萎缩,部分患者单光子发射计算机断层成像(SPECT)发现相应脑区代谢减低,血流减少。例10脑组织病理提示运动皮质、视觉皮质、背侧苍白球和前丘脑有许多微观皮质梗死以及大梗死。6例患者给予帕金森药物治疗,仅例1有改善,余5例症状均缓解不佳。4例患者给予脑血管病二级预防治疗(阿司匹林、他汀类药物等)。最终诊断考虑为皮质基底节综合征(corticobasal syndrome,CBS)7例,脑血管病3例。本例患者诊断为先天性ICA缺失相关的VaP,CBS待排除。
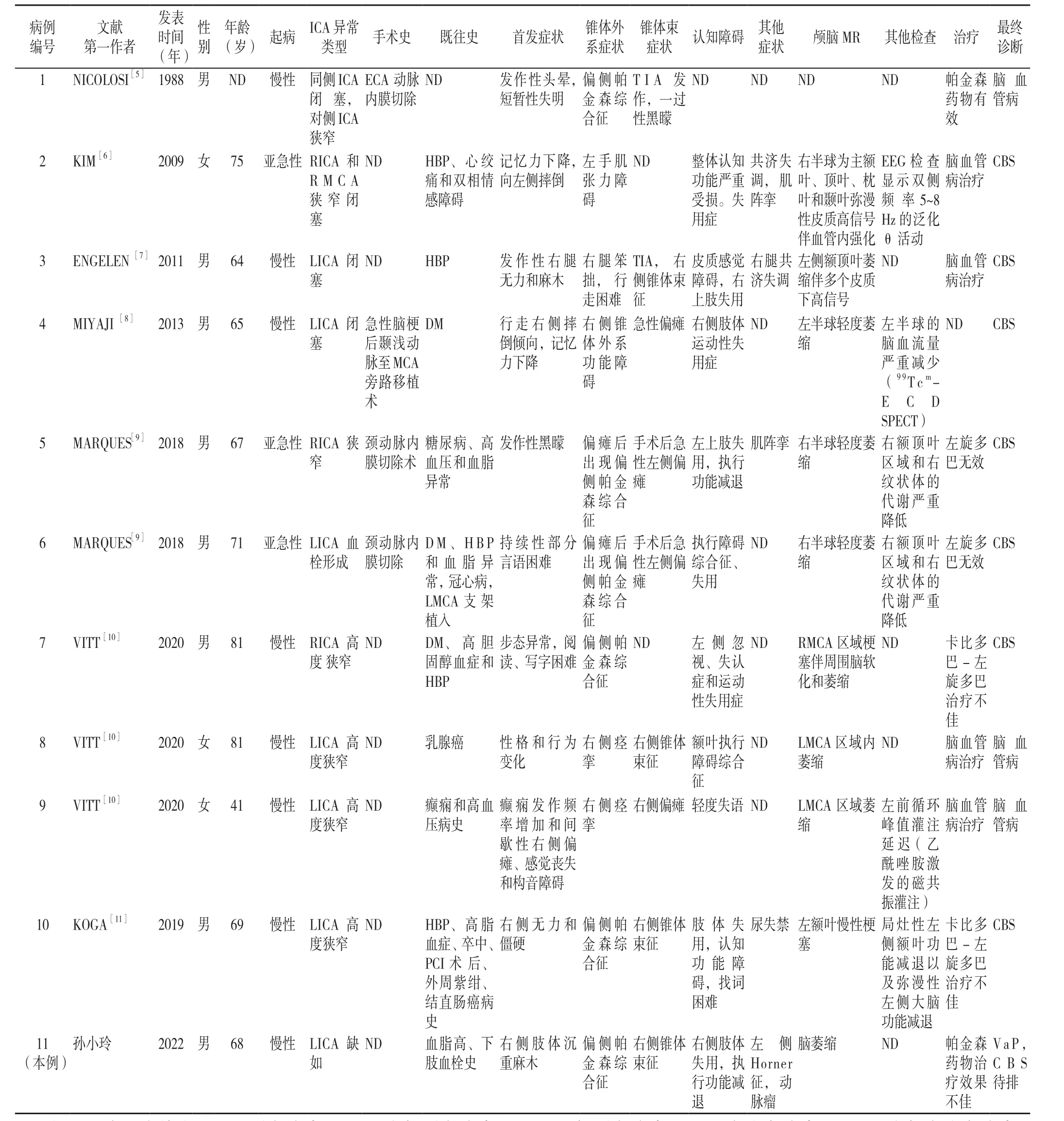
表1 颈内动脉闭塞/狭窄与帕金森综合征的文献复习Table 1 Review of the literature on internal carotid artery occlusion/stenosis and Parkinson's syndrome
3 讨论
先天性ICA缺失是一种罕见的脑血管发育异常。1968年由LIE[12]首次报道了6种侧枝代偿形式的先天性ICA缺失,至今文献报道百例左右。先天性ICA缺失以左侧多见,发病率约为右侧的3倍,也可以累及双侧。ICA异常单独存在时很少引起症状。大多数情况下由对侧 ICA和通过Willis 环的椎基底动脉系统提供血流,或者通过持续的原始通路(即海绵间吻合)[13-14]或来自颈外动脉的经颅侧支循环提供血流。先天性ICA缺失本身不会引起异常症状,但与之相关的其他脑血管血供异常[3]。ZHANG等[2]报道了64例先天性ICA缺失病例,临床症状谱从无症状到先天性Horner综合征、视野缺损或视力下降、搏动性耳鸣、三叉神经痛等,且存在在老年人群中TIA好发、年轻人群中SAH以及发育迟缓常见等特点;该人群中Lie A 型和Lie B型为单侧ICA发育不全中最常见的类型。
本例患者在早年就诊时颈部CTA已显示先天性左侧ICA缺失,伴有左ICA-C7段小动脉瘤以及右锁骨下动脉起始部瘤样扩张,缺如侧大脑前动脉及大脑中动脉均通过前交通动脉由对侧颈内动脉供血,符合Lie B型改变。临床症状、体征表现为左侧先天性Horner综合征,应与颈交感神经发育不良有关。该患者在早期还表现为右侧锥体束征,右侧锥体外系受累的征象。经近7年的随访,发现患者右侧肢体症状逐渐加重,左侧无受累,呈明显的不对称性;还逐渐出现右侧肢体失用与认知功能障碍,常规抗帕金森药物治疗欠佳,因此,考虑该患者的发病机制是在先天性ICA缺失的基础上,血管相关的发病机制所致的VaP。典型的VaP多由动脉粥样硬化引起,壳核苍白球丘脑通路上关键部位的梗死或丘脑皮质通路上弥漫性皮质下白质小血管病变,均可产生“下半身帕金森综合征”[15],表现为双侧对称的步态异常,伴有锥体束征、大小便失禁,对多巴胺药物反应差等。该患者临床经过及表现不符合典型的动脉粥样硬化相关的VaP表现,影像学上也未见相关部位受累的征象,因此推测本例患者的发病机制应与经典的VaP不同。
CBS被归类为非典型帕金森病,以运动和高级皮质功能障碍为特征。CBS与皮质基底节变性(corticobasal degeneration,CBD)相关,但CBD是神经病理学术语,描述了“异常过度磷酸化的4R tau蛋白沉积在皮质、脑干和基底神经节中”的一种病理状态[16]。通过文献复习可发现,在有锥体外系症状、认知障碍和不对称脑萎缩证据的患者中要考虑慢性低灌注和大血管严重狭窄或闭塞的重要性[5-11,17]。推测部分CBS可由ICA闭塞相关的血管所致,ICA闭塞和某些脑区脑血流的连续耗竭引起血流量减少与病理性氧气摄取量增加,引起皮质和基底核神经元网络的慢性病理变化,导致CBS样症状。通过本次报道的病例以及文献复习发现,所有存在明确ICA缺失/慢性ICA闭塞现象的患者,在临床症状上排除锥体束征外,锥体外系症状以及皮质功能损害非常突出,同时影像学也提示相应供血区脑萎缩、代谢减少以及血流量减少,均支持血管发病机制。例10死后脑组织病理也提示脑血管病的改变。2016年ALI等[18]通过PET和SPECT研究进一步证实,CBS患者糖代谢和脑血流减少的分布模式不对称,临床症状较重一侧的对侧额顶叶皮质和皮质下结构(如尾状核和壳核)受影响更大。因此,结合本例患者先天性左侧ICA缺失,渐进性加重的皮质功能(认知功能,失用)和皮质下功能(皮质感觉障碍、肌张力障碍)障碍,10年病程中仍局限于右侧肢体,常规帕金森药物治疗无效,慢性血管机制介导的CBS不能排除,这需要更长时间的随访观察。
综上所述,先天性ICA缺失是一种罕见的脑血管发育异常疾病,可通过常规头颈CTA或MRA筛查确诊,需注意识别其各种神经系统症状,如Horner综合征、搏动性耳鸣、TIA等。但如果同时出现锥体外系症状、认知障碍和不对称脑萎缩证据,需考虑血管机制相关的CBS可能。
作者贡献:孙小玲负责病例收集、文献检索、论文起草;李志军负责最终版本修订,对论文负责。
本文无利益冲突。
猜你喜欢
体育科技文献通报(2022年5期)2022-06-05
中国药学药品知识仓库(2022年6期)2022-04-11
老年医学研究(2021年5期)2022-01-19
天津医科大学学报(2021年4期)2021-08-21
现代仪器与医疗(2021年2期)2021-07-21
世界科学技术-中医药现代化(2021年10期)2021-03-02
心肺血管病杂志(2020年5期)2021-01-14
小资CHIC!ELEGANCE(2021年46期)2021-01-11
睿士(2020年11期)2020-11-16
VOGUE服饰与美容(2020年9期)2020-09-02
