
The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis
2023-02-24 05:24IlariaMartinelliElisabettaZucchiCeciliaSimoniniGiuliaGianferrariGiovannaZamboniMarcelloPintiJessicaMandrioli
中国神经再生研究(英文版) 2023年7期
Ilaria Martinelli ,Elisabetta Zucchi ,Cecilia Simonini ,Giulia Gianferrari,Giovanna Zamboni,Marcello Pinti ,Jessica Mandrioli
Abstract Although mutations in the superoxide dismutase 1 gene account for only a minority of total amyotrophic lateral sclerosis cases,the discovery of this gene has been crucial for amyotrophic lateral sclerosis research.Since the identification of superoxide dismutase 1 in 1993,the field of amyotrophic lateral sclerosis genetics has considerably widened,improving our understanding of the diverse pathogenic basis of amyotrophic lateral sclerosis.In this review,we focus on cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis patients.Literature has mostly reported that cognition remains intact in superoxide dismutase 1-amyotrophic lateral sclerosis patients,but recent reports highlight frontal lobe function frailty in patients carrying different superoxide dismutase 1-amyotrophic lateral sclerosis mutations.We thoroughly reviewed all the various mutations reported in the literature to contribute to a comprehensive database of superoxide dismutase 1-amyotrophic lateral sclerosis genotype-phenotype correlation.Such a resource could ultimately improve our mechanistic understanding of amyotrophic lateral sclerosis,enabling a more robust assessment of how the amyotrophic lateral sclerosis phenotype responds to different variants across genes,which is important for the therapeutic strategy targeting genetic mutations.Cognition in superoxide dismutase 1-amyotrophic lateral sclerosis deserves further longitudinal research since this peculiar frailty in patients with similar mutations can be conditioned by external factors,including environment and other unidentified agents including modifier genes.
Key Words:amyotrophic lateral sclerosis;cognitive impairment;genotype-phenotype correlation;superoxide dismutase 1
Introduction
Amyotrophic lateral sclerosis (ALS) is traditionally defined as a fatal motor neuron disease causing progressive muscle weakness and wasting.In the last decade,fast-growing knowledge about ALS has allowed defining it as a multi system disorder that involves cognitive impairment in at least half of the cases,well beyond the sole motor neuronal system (van Es et al.,2017).
Approximately 10–15% of ALS cases are familial (fALS),while the remaining 85–90% are considered sporadic (sALS).Superoxide dismutase 1 (SOD1)mutations represent the second most common cause of genetic ALS,accounting for approximately 18.9% of fALS and 1.2% of sALS (Zou et al.,2017).
SOD1mutations show a distinct impact on the ALS phenotype across a wide spectrum.Cognitive impairment is not considered a prominent feature of SOD1 ALS (Wicks et al.,2009;Roggenbuck et al.,2017),but recent evidence seems to contradict this assumption.In this focused review,we will examine reports concerning ALS patients with SOD1 mutations with symptoms and signs of cognitive impairment.
We will also discuss the clinical and radiological correlates of cognitive and behavioral impairment in SOD1-ALS.
Search Strategy
We performed a bibliographical search on the PubMed database including all articles published in English up to April 2022.We used the core search terms “amyotrophic lateral sclerosis” or “ALS” and “SOD1” combined with each of the following keywords separately: “cognition”,“cognitive impairment”,“behavioral change”,“dementi a”,“frontotemporal dementi a”,and “neuropsychological testing”.A supplementary search combined the core search terms with the following keywords: “extra-motor”,“PET”,and “MRI”.Two independent researchers evaluated the reviewed articles.We filtered the database search to show original articles referred to as experimental evidence,randomized controlled clinical trials,observational studies,case reports,and case-study series.
Superoxide Dismutase 1 Gene and Protein
The humanSOD1gene is located on chromosome locus 21q22.1 and is composed of five exons separated by four introns (Rosen et al.,1993).The encoded SOD1 protein is a Cu/Zn superoxide dismutase,a 16 kDa homodimer of a 153-amino acid polypepti de.Each SOD1 subunit consists of a β-barrel core and seven loops at the edge which are held together by an intramolecular disulfide bond,a binuclear metal binding site,and a global hydrogen bond network.The metal binding site holds a copper and a zinc ion and is responsible for the catalytic acti vity of SOD1 (Huai et al.,2019).Posttranslational modifications,including full methylation and disulfide bonding,enhance protein stability in the cytoplasm and favor dimerization.These passages are crucial not only for the stability of the SOD1 protein but also to achieve its full catalytic properti es (Sirangelo et al.,2017).
SOD1 is found in all cells in almost alleukarya.Its amino acid sequence is highly conserved through evolution,suggesting that SOD1 plays a crucial function in cellular homeostasis.In eukaryotic cells,SOD1 protein is found ubiquitously in the cytosol,nucleus,peroxisomes,and mitochondrial intermembrane space,where it controls the levels of reactive oxygen species(ROS),mostly produced by the mitochondrial respiratory chain (Peggion et al.,2022).
From a functional point of view,SOD1 acts as a defender from ROS lowering the steady-state concentration of superoxide by converting two superoxide anions (Mathis et al.,2019),which are by-products of cellular respiration with oxidant acti vity,into oxygen and hydrogen peroxide (Azadmanesh et al.,2018).
In 1993,SOD1was the first identified mutated gene shown to cause fALS(Rosen et al.,1993).The discovery of the role ofSOD1in ALS significantly improved our understanding of the disease as it allowed,for instance,the development ofSOD1transgenic animal models of ALS.
Since then,an increasing number of variants inSOD1have been identified in fALS and sALS patients.Currently,more than 200 mutations have been discovered in this gene (http://alsod.iop.kcl.ac.uk/),including single-point mutations,deletions,insertions,and truncation mutations throughout the five exons of theSOD1gene.Missense mutations are the most represented,being widely distributed in the enti re coding region and dominantly inherited(Saccon et al.,2013;Lin et al.,2019).At least ten nearsplice/intronicSOD1variants,affecting allSOD1introns,have been reported in ALS patients(Muratet et al.,2021).Insertions/deletions within theSOD1coding sequence are also frameshift variants identified in ALS patients (Muratet et al.,2021).
The exact mechanism by which these alterations cause ALS remains elusive,oscillating between loss-or gain-of-function mutations.
The loss-of-function mechanism of SOD1 protein was initi ally speculated as the driving force in ALS pathogenesis (Saccon et al.,2013),but subsequent studies derived from animal models contradict this notion (Boillée et al.,2006).Furthermore,the recent description of SOD1 loss-of-function as a new complex neurological disease with a recessive mode of inheritance(Park et al.,2019;Andersen et al.,2019) complicates the issue: a reduction in enzyme activity—as in heterozygous carriers—does not lead to familial ALS (Bali et al.,2017) whereas complete loss-of-function culminate in a nonpure motoneuronal disease.Indeed,various human SOD1 variants lead to a gain-of-function of the gene product SOD1 with SOD1 aggregation (Une et al.,2021) and mitochondrial dysfunction (Jancovic et al.,2021).Proteinaceous inclusions immunoreactive for SOD1 have been found in spinal cords from transgenic mice and ALS-SOD1 patients,and it is generally believed that aggregation of SOD1 plays a key role in the pathomechanism of SOD1-linked ALS (Boillée et al.,2006).
Mutant SOD1 is postulated to undergo oligomerization through the disulfidecrosslinking associated with exposure of the SOD1 structural interior,triggering the onset and progression of the disease (Tokuda et al.,2017).It has been suggested that SOD1 stability could not only be mined by variants but that unidentified factors could be involved in the formation of pathogenic SOD1 conformationsin vivo(Roggenbuck et al.,2017;Berdyński et al.,2022).Moreover,recent evidence from histopathological studies of sALS andSOD1-mutated patients revealed enzymatically dysfunctional and immature conformations of the SOD1 protein in the ventral spinal cord of all ALS cases,irrespectively of the mutational status of the gene,stressing how this protein is crucial to ALS pathogenesis (Trist et al.,2022).
Nonetheless,the mechanism by which aggregation of SOD1 causes motor neuron death is still uncertain.Various hypotheses have been proposed including excitotoxicity,oxidative stress,mitochondrial dysfunctions,and noncell autonomous toxicity,also implicating altered calcium metabolism (Peggion et al.,2022).
Superoxide Dismutase 1-Amyotrophic Lateral Sclerosis: Genotype and Phenotype
The frequency ofSOD1mutations varies from 12% to 23% in patients with fALS and from 0 to 7% in sALS (Kaur et al.,2016),depending on the examined population,with description of de novo mutation for “truly” sporadic cases(Alexander et al.,2002).Data from the ALSoD database (http://alsod.iop.kcl.ac.uk/) showed that,considering ALS as familiar when more than one member of a family is affected,independently from the legacy degree,theSOD1mutation had a percentage distribution in fALS and sALS of 86% and 13%,respectively (Kaur et al.,2016).
It should be noted,however,that the majority of SOD1 variants are outlined from clinical case reports or case series.Thus,conclusive genotype-phenotype correlations cannot be entirely discussed with confidence (Ticozzi et al.,2011).
Moreover,the pathogenic role of single variants has been questioned,since functional evidence or evidence of familial segregation is often lacking or limited,with an important consequence in genetic testing and counseling in clinical practice (Ticozzi et al.,2011).
Validation of the pathogenicity of allSOD1variants is becoming especially prominent due to a therapeutic intervention targeting SOD1 by antisense oligonucleotides or microRNA delivered by adeno-associated viruses to prevent SOD1 accumulation (Miller et al.,2020;Mueller et al.,2020).
The most common mutations portrayed in fALS are G94A,A5V,H47R,and D91A,with G94A being the first mutation studied in a transgenic mouse model (Valentine and Hart,2003).
In ALS patients,only sevenSOD1homozygous variants (L85F,N87S,D91A,L118V,L127S,L145S,and G28_P29del) have been reported unti l now (Saccon et al.,2013;Gagliardi et al.,2022).In four of these variants (D91A,L118V,L126S,G28_P29del),SOD1 acti vity was measured,varying between 25% and 93% of normal levels.According to the gain-of-function theory,a correlation between different enzymatic activity and clinical aspects of the disease was studied but not confirmed (Cleveland et al.,1995).Clinical expression of homozygotic mutation is variable,ranging from the unusually slow progression of D91A and L118V (Saccon et al.,2013) to the aggressive forms with the young onset of L145S and L85F,with variable survival (Alavi et al.,2013;Gagliardi et al.,2022).
The penetrance ofSOD1mutations is variable and linked to the specific mutation involved:SOD1penetrance is almost complete for A5V while being less than 30% at 70 years for ALS patients carrying I114TSOD1mutation (Orrell et al.,1999).
As far as geographic distribution is concerned,SOD1mutations represent the first genetic cause of ALS in Asians (with H47R more represented in Japan–Yamashita et al.,2015).SOD1is only the second most frequent gene involved in European datasets (Zou et al.,2017;Mathis et al.,2019).
In Northern Scandinavia,D91A exists with an allele frequency of up to 2.5%(Andersen et al.,2001),in association with ALS with both autosomal dominant(Prudencio et al.,2009) and recessive inheritance (Pasinelli and Brown,2006).In Europe,other prevalent mutations are L145F in the Balkans (Ferrera et al.,2003),R116G in Germany (Niemann et al.,2004),G42S in Italy (Battistini et al.,2010).
In the United States,the mutation A5V is the most prevalent,representing about 50% of SOD1-ALS patients (Valentine and Hart,2003),whereas the I114T mutation is by far the most common in the United Kingdom,a variant that is highly diverse regarding the age of onset (Yamashita et al.,2015).
Historically,patients with SOD1-related ALS have been reported as generally presenting some distinctive clinical characteristics compared to those with “classical ALS”: longer duration of disease,earlier age of onset,and prominent lower motor neuron (LMN) involvement,usually beginning in the lower limbs (Andersen et al.,2011;Mathis et al.,2019).Data from the ALSoD database (http://alsod.iop.kcl.ac.uk/) show that approximately 95%of SOD1-ALS patients have spinal onset with predominant LMN involvement(Andersen et al.,2011;Lin et al.,2019),and mean age at onset of 48 years old with no different likelihood between male and female.A comprehensive assessment of determinants of ALS phenotypes (Chiò et al.,2020) reaffirms the predilection of LMN involvement in the lower limbs sinceSOD1mutations were found to be associated with the flail leg phenotype (with a 3.5-fold increased frequency),rather than the bulbar phenotype (with a 3.5-fold reduced frequency) as compared to non-carriers/wild-type patients.
In addition to one-to-one correspondence,increasing data from case reports and larger population studies reveal that phenotypes may vary greatly according to specific mutations (Connolly et al.,2020;Li et al.,2016).Therefore,if A5V,H44R,L85V,G86R,N87S,or G94A mutations are associated with an aggressive form of ALS with a survival usually shorter than 3 years,other mutations including G94C,D91A,or H47R are linked with a longer survival (Li et al.,2016).
The emblematic A5V mutation,like A5T (Aksoy et al.,2003),is associated with rapidly progressive LMN signs in the limbs,trunk,or bulbar innervated muscles and early respiratory involvement,with a survival time restricted to 1 to 2 years (Cudkowicz et al.,1997;Bali et al.,2017).
On the other hand,SOD1recessive mutation D91A is described as associated with a phenotype characterized by a long premotor phase involving the lower back,hip,or knee pain,muscle cramps,and sti ffness in the legs that last for months or even years,followed by the insidious onset of monolateral paresis that progresses slowly to other spinal districts,with an average overall survival of a decade (Pasinelli and Brown,2006).Between these two extremes are the cases of ALS patients heterozygous for the D91A mutation (Parton et al.,2002),who may have variable ALS phenotypes with bulbar or spinal onset,as well as short or intermediate survival.
A shorter survival in SOD1 subjects has been related to a higher propensity for the SOD1 protein to aggregate (Prudencio et al.,2009) or lower its stability(Goutman et al.,2018).Mutations inSOD1can destabilize protein’s structure,acting as a trigger for protein misfolding,the appearance of toxic species,and thus shorter survival times (Goutman et al.,2018).Recently,the aggregation power of A5V mutation on aberrant folding intermediates of SOD1 has been confirmed as a crucial source of cytotoxic conformations in ALS pathology(Berdyński et al.,2022).
Another point regards motor neuron involvement: it is commonly accepted that SOD1-ALS presents predominant LMN features,although upper motorneuron manifestations may also be present,as recently confirmed by Connolly et al.(2020) in a systematic review.45.2% of SOD1-ALS cases revealed an involvement of upper and lower motor neuron,compared to 47.6% with pure LMN signs (Connolly et al.,2020).Accordingly,pathologic MEPs with greatly prolonged central conduction latencies have been described in patients with different mutations (Stewart et al.,2006).
The biological reason behind the predominant LMN features has not been clearly ascertained yet,but animal studies suggest that SOD1 pathology begins at the periphery and proceeds in a retrograde manner (Liu et al.,2015),indicating thatSOD1mutations may make the distal nerve more prone to degeneration (Connolly et al.,2020).
Despite the above-described genotype-phenotype correlations,there remains considerable phenotypic variability in ALS patients carryingSOD1gene mutations (Figure 1andAdditional Table 1),with heterogeneity between and within families.This heterogeneity supports the hypothesis that a single mutation is not the only factor affecting the presentation and clinical course of the disease,but other mechanisms,ranging from genetic background to environment,might also contribute to modulating the phenotype.Armitage-Doll multistep models of ALS demonstrate thatSOD1defects may increase suscepti bility to ALS development by reducing the number of steps from six,found in the wild-type population,to two (Chiò et al.,2018).
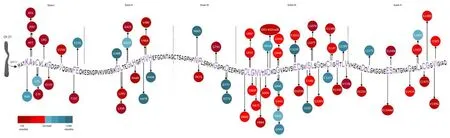
Figure 1|Representation of SOD1 mutations with different prognostic impacts.
Cognitive and Behavioral Impairment in Amyotrophic Lateral Sclerosis
Both in SOD1-ALS and in non-SOD1-ALS,the clinical spectrum of the disease has been enriched by increasing evidence about the involvement of further neuronal domains.Since 2002,literature proven ALS patients can develop cognitive and behavioral manifestations of frontotemporal dementia(FTD),such that today the most appropriate definition refers to the ALSfrontotemporal spectrum disorder (Strong et al.,2017).
In half of the patients along this spectrum,ALS is accompanied by non-motor symptoms (Strong et al.,2017) including variable cognitive and behavioral impairments (Consonni et al.,2021),which emerge through dedicated neuropsychological batteries (Pender et al.,2020).The most frequently reported deficits of cognitive impairment in amyotrophic lateral sclerosis involve executive functioning (e.g.,working memory,verbal fluency,attention,and impulse control),social cognition (e.g.,emotion recognition,“theory of mind task”) and language (e.g.,naming,word and sentence comprehension,speech fluency) (Beeldman et al.,2016).Mild behavioral impairment in amyotrophic lateral sclerosis may affect up to 60% of ALS patients.The most frequently reported deficits of behavioral impairment in amyotrophic lateral sclerosis include apathy,disinhibition,irritability,emotional blunting,abnormal eating behavior,perseveration,and loss of insight (Consonni et al.,2021),as well as psychiatric symptoms such as psychosis (delusions and hallucinations) and mood changes (Zucchi et al.,2019).When these symptoms are severe enough to compromise the cognitive functioning of the patient,beyond the limitations due to motor symptoms,a diagnosis of dementia can be given.In 5–15% of ALS patients,such cognitive and behavioral symptoms are clinically evident and sati sfy FTD diagnostic criteria (Strong et al.,2017).Cognitive manifestations in ALS are more frequent when bulbar functions are involved (Chiò et al.,2019).Motor and cognitive symptoms can worsen simultaneously or separately,but cognitive impairment worsens with disease progression,becoming more frequent in the most severe stages of ALS disease (Chiò et al.,2019).
The disease timeline is still a matter of debate: dementia can precede or develop after the appearance of motor symptoms,developing a typical profile of behavioral variant of FTD.In contrast,patients with bvFTD may eventually display signs of motor neuron involvement without evolving to complete ALS.In some rare cases,there is a parallel,concomitant development of both ALS and bvFTD phenotypes at the same time (Beeldman et al.,2016).ALS-FTD patients have a worse prognosis and shorter survival rates than patients with ALS or bvFTD alone;behavioral and personality disturbances are negative prognostic factors (Strong et al.,2017).Recent prospective cohorts of ALS patients highlighted that FTD-onset patients characteristically present at an older age,with the bulbar onset and signs of upper motor neuron dysfunction.Of relevance,survival between FTD-and motor-onset ALS patients is similar and negatively affected by the appearance of bulbar signs(Gromicho et al.,2021).Early detection and careful monitoring of cognitive deficits in ALS are crucial for patient and caregiver support and enable personalized management of the individual patient’s needs.
Cognitive/Behavioral Impairment and Amyotrophic Lateral Sclerosis-Related Genes
After the first description of transactive response DNA-binding protein 43(TDP-43) as the underlying pathology of the ALS-FTD spectrum,mutation of the corresponding gene (TARDBP) was found in about 1.2% of sALS and 3% of fALS (Zou et al.,2017).TDP-43,the major disease protein in up to 97% of ALS with or without dementia (Mackenzie and Rademakers et al.,2008),is a nuclear protein that in ALS mislocalizes in the cytoplasm.Its identification as the pathological signature protein in FTD with ubiquitinpositive inclusions (FTD-TDP) and in ALS has further consolidated the concept of its cardinal involvement in ALS pathophysiology (Geser et al.,2010).Patients carrying TARDBP mutations may present with (or develop during the disease) cognitive impairment,particularly in the executive and linguistic domains (Floris et al.,2015),often consistent with the temporal variant of FTD or semantic dementi a (Spinelli et al.,2022).A large cohort ofTARDBPmutated patients with motor neuron disease was studied by quantitative MRI advanced techniques (Spinelli et al.,2022) showing a distinctive parietal pattern of cortical atrophy and greater damage of motor and extra-motor tracts compared with matched non-TARDBP ALS,along with a lower linguistic performance at neuropsychological tests.
A molecular link between motor and non-motor symptoms was further confirmed with the identification of a repeat expansion within theC9orf72gene (Renton et al.,2011) linking FTD and ALS.Neuropathological investigations have also shown that motoneurons withC9orf72expansions are characterized by TDP-43 accumulation and mislocalization in specific neuroanatomical regions (Balendra and Isaac,2018;Breevort et al.,2022).C9orf72hexanucleotide repeat expansions are the most common cause of familial FTD-ALS worldwide.The clinical presentation is often indistinguishable from classic FTD or ALS,although neuropsychiatric symptoms are more prevalent and,for ALS,behavioral and cognitive symptoms occur more frequently than in cases of semantic dementia and progressive non-fluent aphasia (Breevort et al.,2022).Behavioral features have been described in cohorts of patients with FTD andC9orf72expansion (C9FTD) and less frequently in C9FTD/ALS patients.C9orf72expansion carriers may have an atypical neuropsychiatric presentation involving associated hallucinations or delusions and a greater risk of psychiatric disorders (Zucchi et al.,2019).If more than half of C9FTD subjects develop motor neuron symptoms throughout the disease (Mahoney et al.,2012),approximately 50% of C9ALS subjects develop cognitive impairment during the disease,with bvFTD as its most common presentation,followed by impairments in executive domains and apathy.Disinhibition,apathy,compulsivity and loss of empathy were noted in a small percentage (Chiò et al.,2012;Floeter et al.,2017).
Adjunctive findings involving protein recycling and disposal have pointed out the pathological convergence between ALS and ALS-FTD.For example,ubiquilin 2 (UBQLN2),optineurin (OPTN),SQSTM1/p62,and valosincontaining protein (VCP)—all related to protein degradation pathways—have been linked to both diseases (Shahheydari et al.,2017).Ubiquilin-2 positive inclusion pathology was found not only in patients withUBQLN2mutations but also in their sporadic counterparts.The abnormal presence of positive Ubiquilin-2 inclusion in the cytosol of degenerating motor neurons of fALS and sALS patients has been recently linked to neurodegeneration (Renaud et al,2019).Patients carrying theUBQLN2mutation can manifest a progressive dementi a with frontotemporal impairment,including abnormalities in both behavioral and executive functions (Ranganathan et al.,2020).
VCP is an ATPase involved in the degradation of abnormal proteins,specifically in targeting endoplasmic reticulum proteins for degeneration(Shahheydari et al.,2017).Johnson et al.(2010) linked mutations in theVCPgene to several families with typical ALS,and with individuals presenting FTD.Mutations in theVCPgene were already known to underlie an unusual clinical syndrome characterized by inclusion body myopathy,Paget’s disease of the bone and FTD (Ranganathan et al.,2020;Scarian et al.,2022).Mehta et al.(2013) studied genotypic-phenotypic relations in 27 families,grouping them according to VCP variants with different phenotypes;VCP-related ALS and FTD were phenotypically indistinguishable from sporadic forms except for a younger onset in VCP-related FTD (Mehta et al.,2013).
Fused-in-sarcoma (FUS) is a member of the hnRNP family,located on chromosome 16p11.2,a chromosomal region linked to multiple fALS cases(Vance et al.,2009).Mutations in theFUSgene can cause ALS and are associated with rare case reports of patients with clinical FTD,without the description of pathology (Deng et al.,2014).An accumulation of FUS protein in inclusion bodies of neuronal cytoplasm and nucleus has been associated with clinicopathological subtypes of FTD (Perry et al.,2017),with TDP-43 and FUS pathology being mutually exclusive (Vance et al.,2009).Exploring clinicopathological correlations in a large bvFTD cohort,Perry et al.(2017)found that patients with frontotemporal lobar degeneration associated with FUS were younger and had more severe behavioral impairment than all other bvFTD groups.In addition to the core bvFTD atrophy pattern,severe ventromedial frontal,anterior temporal,and striatal volume loss separated this group from others (Perry et al.,2017).Charged Multivesicular Body Protein 2B (CHMP2B) gene mutations are characterized by perturbations to normal endosomal-lysosomal and autophagic trafficking,resulting in the aberrant accumulation of enlarged endosomes and autophagic organelles(Zhang et al.,2017).Although mutations inCHMP2Bare rare,the importance of CHMP2B to the FTD-ALS spectrum lies in the distinctive pathology observed,with TDP-43 negative,ubiquitin and/or p62 positive inclusions(Ugbode and West,2021).Affected individuals typically present in their late 50’s with a behavioral variant of FTD (Skibinski et al.,2005).
Finally,TBK1(TANK Binding Kinase 1) is the gene coding for a serine/threonine kinase that plays a crucial role in autophagy (Kim et al.,2020)leading to the accumulation of TDP-43,and commonly resulting in an FTDALS syndrome,sometimes with atypical features.Being at the intersection of neuroinflammation and protein quality control,TBK1 exerts antiviral and antibacterial response under activation of Toll-Like receptor signaling pathways,but also has a significant role in autophagy and mitophagy,chiefly the phosphorylation of autophagy adaptors (Oakes et al.,2017).From a clinical point of view,TBK1 mutations have been linked to various cognitive and motor expressions: recently,Swiftet al.(2021) revised the clinical phenotype of TBK1 mutation carriers,finding that 58% of cases presented with ALS and 16% with FTD-ALS overlap.Among them,the FTD phenotype was described in 12 subjects (6 with bvFTD,3 with non-fluent,and 3 with semantic primary progressive aphasia).
Cognitive Impairment in Superoxide Dismutase 1-Amyotrophic Lateral Sclerosis
Literature data have depicted the classical view of “cognitively spared” ALS patients with SOD1 mutation (Wicks et al.,2009),especially in comparison to non-SOD1 fALS patients (Marjanovic et al.,2017) likeC9orf72expanded cases(Renton et al.,2011).This might be explained by the fact that classical SOD1-ALS patients typically have a younger age at onset and a spinal phenotype with lower motor neuron predominance,all phenotypic traits less commonly found in ALS-FTD patients (Gromicho et al.,2021).
In line with this,neuroimaging data support the sparing of cognitive circuits in SOD1-ALS patients.Indeed,Agosta and colleagues investigated structural and functional MRI alterations of the brain using a sample of 20 SOD1-ALS patients compared to 33 controls,revealing no significant cortical thinning or increased functional connectivity of the precentral cortex of SOD1-ALS patients.In contrast,analysis of structural MRI data of the cervical cord showed significant cord atrophy in patients with SOD1-ALS compared to those with sporadic ALS,which correlated with disease duration and functional impairment (Agosta et al.,2018).
However,more recent single-case reports and case series undermine the hypothesis that SOD1-ALS derives from a distinct neurodegenerative pathway that spares frontotemporal functioning (Additional Table 2).
Accordingly,a possible role of SOD1 gain-of-function in cognition was hypothesized in Down Syndrome,characterized by an extra copy of chromosome 21 and by early onset Alzheimer dementia.Together with a role of theAPPgene,a possible role of theSOD1gene has also been suggested in this gene dosage disease where the overproduction of ROS,enhanced oxidative damage,and possibly ROS-induced cell death has been demonstrated,along with an aberrant expression of SOD1 on human brain studies (Lanzillotta and Di Domenico,2021).
In SOD1-ALS patients,the most specific characteristics of cognitive impairment suggest the involvement of the frontal lobes and fall within the ALS-FTD spectrum,with only a few exceptions (Additional Table 2).To mention a few,Muller et al.(2018) reported a patient with a SOD1 H49R mutation presenting with aggression,emotion lability,reduced working memory,and slightly reduced verbal fluency,suggestive of an initial bvFTD presentation with an abnormal off-key liquoral analysis,and consistent with Alzheimer’s disease biomarker profile.
Battistini et al.(2010) described an Italian patient with ALS secondary to a G42S mutation who manifested sexual and urinary disturbances,visual hallucinations,and mental confusion,and whose clinical cognitive and behavioral profile was not enti rely consistent with typical bvFTD.
BvFTD was also the clinical diagnosis assigned to cases described by other Italian reports.Indeed,vulnerability in other non-executive cognitive circuits has been suggested in an fALS case carrying G148C mutation,with alterations in the right supramarginal gyrus at brain imaging;the clinical follow-up along with neuropsychological tests and 18F-FDG PET showed a progressive trend toward cognitive impairment with frontal lobe involvement (Canosa et al.,2014).In a family of ALS with an L145F mutation,SPECT images confirmed a hypoperfusion in both frontal regions,especially on the left side and on the left temporal cortex,along with a right-crossed cerebellar diaschisis (Masè et al.,2001).
Extrapyramidal involvement was reported in patients with ALS and chorea manifestations (Lopate et al.,2010) or hypothalamus dysfunction (Nakamura et al.,2014).Surprisingly,in a case-series of early-onset dementi a patients with unclear phenotypes (Perrone et al.,2018),SOD1I114T mutation was identified in a patient with an initi al diagnosis of Korsakoff syndrome without motor impairment.A first detailed case of fulminant dementia associated with the same mutation (I114T) was reported by Katz,describing a patient with changes in emotions,personal conduct and self-awareness,and subsequent severe aphasia;a cerebral MRI confirmed involvement of frontal and temporal regions with selective atrophy (Katz et al.,2012).
Heterogeneous phenotypic manifestations of specificSOD1mutations have been observed even in the same family: for example,the truncating mutation G142X (Nakamura et al.,2014) was described in probands with FTD and later progressive motor neuron disease,while all other family members had earlyonset and rapidly progressive ALS without cognitive impairment.Cognition and behavior may be affected early in the disease course (Martinelli et al.,2020) or in its later stages,worsening with more severe disease stages(Crockford et al.,2018).
Interestingly,Glycine substitutions at 42 codon were reported by distinct groups (Battistini et al.,2010;Niu et al.,2016) of ALS patients with frontal lobe dysfunction and behavioral symptoms,suggesting the possibility of a specific point of structural liability in the protein,which is more frequently linked to extra-motor manifestations.
From these case series of cognitively impaired SOD1-ALS patients,it is difficult to establish whether cognition and behavior are an expected target of the long-lasting degenerative process,initiating in the motor cortex and slowly spreading to the prefrontal cortex,thus fully manifesting only in the slowly progressive cases (whereas it may never become evident in the more aggressive forms).Cognitive/behavioral impairment may occur as an independent,parallel pathology resulting from the same genetic and environmental triggers of the motor manifestation of ALS.SOD1 murine models may shed light on peculiar patterns of spati al-temporal progression and offer a biological parallel to some clinical observations.For example,Synofzik found a mild cognitive impairment in a patient carrying the G94A mutation (Synofzik et al.,2010),discussing its similarity to rodent G94A models where abnormal prefrontal cortex connecti vity and function herald the onset of motor disturbances (Sgobio et al.,2007).Similarly,Filali et al.(2011) reported data on perseverant behavior in the G38R SOD1 transgenic mouse model,attributable to prefrontal connectivity dysfunction,while impairments in passive avoidance learning have been pustulated (Spalloni et al.,2016),along with a progressive degeneration in the motor functions.
There are only a few studies that have directly compared SOD1-ALS patients with non-mutated ones on extended neuropsychological batteries,with contrasting conclusions.Wicks et al.(2009) found that individuals withSOD1gene mutations do not have significant differences in their neuropsychological performance compared to non-SOD1 ALS.Marjanovic et al.(2017) however,demonstrated that SOD1-ALS patients mainly present a selective dysexecutive syndrome,whereas SOD1 negative patients present a more widespread cognitive impairment including—in addition to executive dysfunction—visuospati al and memory deficits,suggesting the involvement of both frontal and posterior cortical regions.
Finally,functional brain alterations by PET images were collected from SOD1-ALS patients and asymptomatic SOD1-carriers,speculating that this might reflect differences in cortical neuronal vulnerability.
The first data collection came from Turner et al.(2005) who applied GABA-A receptor ligand [11C] flumazenil PET in patients carrying homD91A mutation,including two pre-symptomaticSOD1homozygotic D91A carriers,compared to non-mutated ALS patients.This study provided evidence for differences in metabolic patterns: in the non-mutated group,PET hypometabolism was found within premotor regions,motor cortex and posterior motor association areas,while in the homozygotic D91A ALS patients,hypometabolism was predominant in the left frontotemporal junction and anterior cingulate gyrus.Interestingly,in the two pre-symptomatic D91A subjects,a small focus of reduced [11C] flumazenil binding at the left frontotemporal junction was found,like the pattern seen in the clinically affected patients.
Subsequently,a PET study including asymptomaticSOD1mutation carriers and SOD1-ALS patients reported an increased uptake of a microglia activation tracer (11CPK11195) in the motor cortex of symptomatic subjects (Tondo et al.,2020).More specifically,both the symptomatic and asymptomaticSOD1-carriers showed significant microglia activation in cortical and subcortical structures,with clusters of activation in occipital and temporal regions,cerebellum,thalamus,and medulla oblongata,that showed variable patterns at an individual level.SOD1-patients also showed microglia activation in supplementary and primary motor cortices,as well as the somatosensory regions.The authors hypothesized that microglial activation,together with astrocyte reaction,might be the phenomenon underlying glucose hypermetabolism that has been extensively reported in ALS.More recently,Canosa et al.(2022) explored the metabolic differences detected with 18F-FDG-PET in SOD1-and non-mutated ALS patients along with healthy controls,finding a relative hypermetabolism in the motor cortex of SOD1-ALS patients compared with sporadic ALS and healthy controls.
Conclusion
The discovery of theSOD1gene has been crucial for ALS research,as it led to the first mouse model of the disease and provided the basis for numerous pathophysiologic investigations (Goutman et al.,2018).
Since its first identification in 1993,the field of ALS genetics has greatly expanded in multiple directions.The discovery of disease-causing and disease-modifying genes has improved our understanding of the diverse pathogenic basis of ALS.Nonetheless,previous literature data challenge the notion of a strict genotype-phenotype correlation and support the idea that disease expression in the same genetic background can be influenced by external factors.More data are required to further characterize the complex mechanisms of each mutated gene product and their interaction at the cellular level,as well as with the environment.
In this review,we aimed to outline the SOD1-ALS clinical framework with a focus on cognitive impairment.Classical literature has reported that SOD1-ALS forms are cognitively spared;however,more recent reports demonstrate frontal lobe frailty in patients carrying different SOD1-ALS mutations.
Cognitive and behavioral symptoms in SOD1 ALS deserve further longitudinal research,which will improve our mechanistic understanding of ALS by enabling a more robust assessment of how the ALS phenotype correlates to different variants across multiple SOD1 mutations.
In turn,studying ALS genotype-phenotype correlations among the different mutations will contribute to a comprehensive database of these correlations.Since therapeutic strategies are upcoming for SOD1-ALS (Kim et al.,2020;Miller et al.,2020;Mueller et al.,2020),the study of phenotypic heterogeneity associated with SOD1-ALS is even more important.Indeed,interpreting the pathogenicity of allSOD1variants is a crucial passage for the development of advanced therapeutic intervention targetingSOD1,thus preventing its accumulation.
Author contributions:Conceptualization: IM,JM;methodology: IM,EZ;writing—original draft preparation: IM,EZ,GG,CS,JM;writing—review and editing: IM,GG,GZ,MP,JM;supervision: GZ,JM,MP.All authors have read and agreed to the published version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Cinzia Volonté,National Research Council (CNR),Italy.
Additional files:
Additional file 1:Open peer review report 1.
Additional Table 1:Systematic collection of literature data on SOD1-ALS patients.
Additional Table 2:Mutations of the SOD1 gene found in ALS patients with cognitive impairment.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- CMT1A current gene therapy approaches and promising biomarkers