
合成气制低碳烯烃串联反应中Zn-Al 氧化物的制备及性能
2023-02-27 07:35李保珍孟凡会王丽娜
燃料化学学报 2023年1期
李保珍 ,孟凡会 ,王丽娜 ,李 忠
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室, 山西 太原 030024)
低碳烯烃( C=2−C=4,乙烯、丙烯和丁烯)是有机化工中最重要的基础原料,可通过石脑油蒸汽裂解、烷烃脱氢、合成气直接或间接转化等方法制备[1]。中国能源结构的基本特点是“富煤、贫油、缺气”[2],发展煤基合成气(CO+H2)制低碳烯烃有助 于实现煤炭资源清洁高效利用,缓解能源危机,具有广阔的工业应用前景[3]。相比于合成气经甲醇间接制备低碳烯烃技术[4],合成气一步法直接合成低碳烯烃具有流程短、能耗低等优势,成为目前的研究热点[5]。近年来,世界各国研究者已经开发出一系列基于C−O 活化和C−C 耦合策略的双功能催化剂催化合成气制低碳烯烃路线(STO)[6−11],能够突破费托合成路线的Anderson-Schulz-Flory(ASF)产物分布规则[12],实现低碳烯烃高选择性[13,14]。
在双功能催化剂(OX-ZEO)中,CO/H2首先在金属氧化物上活化并形成甲醇和/或二甲醚[15]或烯酮[16]中间体,活性中间体迅速扩散进入酸性分子筛择形催化生成低碳烯烃[17,18]。由于CO/H2活化并生成活性中间体是整个催化反应的速率控制步骤[19],且均发生在金属氧化物表面上,因而金属氧化物决定了反应的催化活性。一般来说,适用于OX-ZEO 的金属氧化物组分需具备双活性中心:一是活化CO 活性中心;二是可以解离活化H2促进CO 加氢生成含氧中间体的活性中心。ZnO 可以实现对H2的异裂解离[20],掺杂Cr[21]、Al[22]、Mn[23]等元素可调控表面氧空位含量和H2解离能力,协同活化CO 和H2,促进CO 加氢反应活性。例如,ZnCrOx氧化物耦合SAPO-34 分子筛用于STO 反应时,在CO 转化率达到17%时−C=4的选择性高达80%[6]。研究还发现,ZnAl2O4尖晶石上Zn 和Al 位点可有效协同催化CO 加氢反应生成甲醇和二甲醚[24];与SAPO-34 复合后制备的双功能催化剂用于STO 反应时,Zn-Al/SAPO-34 表现出较好的低碳烯烃选择性[25]。在相同的制备和评价条件下,对比Zn-Al、Zn-Cr、Zn-Zr 和Zn-Ce 氧化物复合SAPO-34 分子筛的催化性能发现,Zn-Al/SAPO-34 表现出更好的催化性能[26]。尽管关于Zn-Al 氧化物活化CO/H2的研究已有大量相关文献的报道,但Zn-Al/分子筛组成的双功能催化剂催化STO 反应的CO 转化率仍有待进一步提高[26−28]。
Zn-Al 氧化物的制备方法较多[29],不同制备方法直接影响氧化物催化反应的构效关系[30]。根据锌铝酸盐晶体形成和生长机制[31]可知,晶体缺陷的存在有利于离子跨氧化物界面的传质,对介孔金属铝酸盐的相形成和晶体生长起着重要作用。因此,通过增加载体中的缺陷,改善原子在界面的传质,可促进金属离子的扩散和进一步插入,有利于新晶相的形成。Fulvio 等[32]以拟薄水铝石为前驱体,通过微波加热制备了孔径为16 nm 的γ-Al2O3比传统加热条件下获得的氧化铝具有更多的酸性和碱性位点。Gonçalves 等[33]通过适当修改上述合成方法,在低温下制备出了具有较大孔径的MeAl2O4(Me= Ni、Co or Cu)尖晶石结构,有助于气体扩散和反应中间产物的快速转移。
本研究以工业薄水铝石为铝源,通过微波辅助蒸发诱导自组装(M-EISA)法,制备不同Zn/Al原子比的Zn-Al 氧化物,并复合SAPO-18 用于合成气直接转化制低碳烯烃。采用XRD、TEM、BET、TPD、XPS、TPR 等对Zn-Al 氧化物进行了表征分析。并与浸渍法制备的Zn-Al 氧化物催化剂进行了对比,研究了制备方法及Zn/Al 原子比对氧化物结构及物化性质的影响,并讨论了催化剂的构效关系。
1 实验部分
1.1 催化剂的制备
采用M-EISA 法[34]制备Zn-Al 氧化物。以Zn/Al原子比为1∶2 的氧化物为例,首先在40 mL 无水乙醇中溶解5.00 g 的三嵌段共聚物P123(平均相对分子质量为5800),然后加入7.93 g 六水硝酸锌(99.0%,国药集团),室温下搅拌4 h 得到溶液A。将2.40 g 拟薄水铝石(AlOOH,中石化大连石油化工研究院)溶于30 mL 去离子水并滴加0.52 mL 浓硝酸得到溶液B,然后在70 ℃微波辐射下搅拌(300 r/min)1 h 形成白色半透明的铝凝胶,冷却至室温后与溶液A 混合并继续搅拌4 h,混合物先后在40 和80 ℃下各真空干燥48 h,最后在马弗炉中500 ℃(1 ℃/min)焙烧4 h 得到的氧化物记为ZnAl2Ox。采用同样方法制备Zn/Al 原子比为2∶1、1∶1、1∶3 和1∶5 的氧化物,并分别记为ZnAl0.5Ox、ZnAl1Ox、ZnAl3Ox和ZnAl5Ox。作为对比,将硝酸锌和AlOOH 粉末在马弗炉中500 ℃焙烧4 h,得到单一氧化物ZnO 和Al2O3,其中,以Al2O3为载体浸渍制备的Zn/Al 原子比为1∶2 的氧化物标记为ZnAl-IP。
采用水热法合成SAPO-18 分子筛[35]。所用原料为异丙醇铝(98.5%,上海阿拉丁科技股份有限公司)、磷酸(85.0%,上海阿拉丁科技股份有限公司)、硅溶胶(Sigma Aldrich 公司)以及四乙基氢氧化铵(TEAOH,35%,Alfa Aesar 公司)。首先配制化学计量比SiO2∶ Al2O3∶ P2O5∶ TEAOH ∶ H2O 为0.2 ∶ 1.0 ∶ 1.2 ∶ 2.0 ∶ 40 的溶液并在室温下强烈搅拌2 h,混合物在装有聚四氟乙烯内衬的晶化釜中170 ℃晶化72 h,所得固体经离心、洗涤、110 ℃干 燥12 h 后 在 马 弗 炉 中550 ℃焙 烧6 h 以 得到SAPO-18 分子筛。
将Zn-Al 氧化物与分子筛按质量比2∶1 在玛瑙研钵中研磨混合后,进行压片并粉碎制成20−40 目颗粒的双功能催化剂。
1.2 催化剂的表征
金属氧化物的晶体结构在日本理学SmartLab SE 型X 射线衍射仪上进行,使用CuKα 辐射源(40 kV, 40 mA),扫描5°−80°,扫描速率为10(°)/min。原位X 射线衍射(in-situXRD)测试在安东帕原位池中进行,测试时通入H2/N2气体,并以10 ℃/min进行升温,同时记录样品在不同温度和在400 ℃下保持不同时间的衍射谱图。
采用日本电子株式会社JEOL JEM-2100 场发射透射电子显微镜(TEM)在200 kV 下观察样品的形貌。测试前,先用超声波将样品分散在乙醇中,然后滴加在铜网上自然晾干后进行分析。
样品的BET 测试在北京贝士德3H-2000PS 1/2仪器上进行。测试前,样品先在250 ℃脱气4 h,然后在液氮条件下(−196 ℃)进行N2吸附-脱附实验。
采用美国Micromeritics Autochem Ⅱ 2920 化学吸附测试样品的H2还原性能(H2-TPR)。将50 mg左右的样品置于U 形石英管中,在He 气氛300 ℃下脱气处理1 h,降至50 ℃后,通入10 %H2/Ar 混合气体,待TCD 信号平稳后程序升温至800 ℃,采用热导率检测器(TCD)记录H2消耗量。
金属氧化物的CO 程序升温脱附性能(COTPD)在Autochem Ⅱ 2920 化学吸附仪上进行。将100 mg 左右的样品先在10%H2/Ar 气氛下400 ℃还原处理2 h 后,在He 气氛中降温至50 ℃,然后在15%CO/He 混 合 气 体 中 吸 附30 min 并 用He 吹 扫至基线平稳,最后程序升温至设定温度并用TCD记录CO 脱附信号。H2-TPD 与CO-TPD 测试条件相同,只是将15%CO/He 吸附30 min 改为10%H2/Ar吸附1.5 h,载气换为Ar。
X 射线光电子能谱(XPS)在赛默飞ESCALAB 250Xi 仪 器 上 测 试,仪 器 装 有AlKα 射 线(hv=1486.6 eV),以标准C 1s结合能(284.6 eV)校准各元素的结合能。
采用布鲁克ELEXSYS E500 型电子顺磁共振仪(EPR)分析金属氧化物中的体相氧空位含量。固体粉末称重后置于核磁管中(9.85 GHz,2.0 mW)于常温下测量。
1.3 催化剂的活性评价
在装有石英玻璃管(内径d= 6.0 mm)的高压固定床反应器中测试合成气直接转化制低碳烯烃反应性能。取0.4 g 双功能催化剂置于石英管中并在催化剂上下端装填石英棉,通入H2/CO/N2物质的量比为6∶3∶1 的原料气,在400 ℃、3.0 MPa、4500 mL/(gcat·h)条件下进行评价。反应后产物经冷凝后在Agilent 7890A 气相色谱仪上进行在线分析。采用火焰离子化检测器(FID)和HP-Al/S 色谱柱分析碳氢产物C1−5;采用TCD 和Porapak-Q、HPPLOT/Q 和HP-PLOT 分子筛三根色谱柱串联分析CO、CO2和N2,氮气为内标气。反应后产物碳平衡在95%以上。CO 转化率、C1−5碳氢化合物摩尔选择性、CO2摩尔选择性和时空产率(STY)的详细计算过程可参见本组之前的研究[36]。
2 结果与讨论
2.1 Zn-Al 样品的结构及形貌
采用XRD 分析Zn-Al 氧化物的晶体结构,结果见图1。图1(a)显示,单一Al2O3在45.6°和66.5°处出现微弱的γ-Al2O3特征衍射峰(PDF #29-0063)。采用M-EISA 法引入Zn 制备的ZnAl5Ox氧化物出现弱的ZnAl2O4尖晶石特征峰(PDF #71-0968),未出现Al2O3特征峰,这可能是因为Al 物种以无定型Al2O3相存在。当Zn/Al 原子比为1∶3 时,ZnAl3Ox氧化物还在31.7°、34.4°、36.2°和47.5°处出现了微弱的六方晶系ZnO 特征峰(PDF #36-1451),且随着Zn/Al 原子比的增加,ZnO 的特征峰逐渐增强。这表明,引入的Zn 与Al 先形成ZnAl2O4尖晶石结构,随Zn 含量的增加,过量的Zn 物种以ZnO 晶体结构出现。与ZnAl2Ox相比,采用浸渍法制备的ZnAl-IP 样品具有明显的ZnO 特征峰,Al2O3的衍射峰向低角度略有偏移,这是因为部分Zn 进入Al2O3晶格导致晶格间距膨胀。该结果也说明,ZnAl-IP 中的Zn 和Al 物种之间相互作用较弱,主要以ZnO 和Al2O3形式存在,而微波法制备样品时可使过渡金属前驱体插入氧化铝载体结构中,达到原子水平的高度分散[33]。
为探究代表性ZnAl2Ox氧化物在不同温度和不同气氛下的结构变化规律,采用原位XRD 进行表征。图1(b)显示,在H2气氛下,当温度从30 ℃升高至400 ℃时,ZnAl2Ox氧化物的晶相没有发生明显的变化;且在400 ℃下H2气氛下处理3 h 后,样品的晶相结构也未发生改变,说明ZnAl2Ox氧化物具有良好的热稳定性,处理温度和时间不会明显影响ZnAl2Ox氧化物的尖晶石结构。
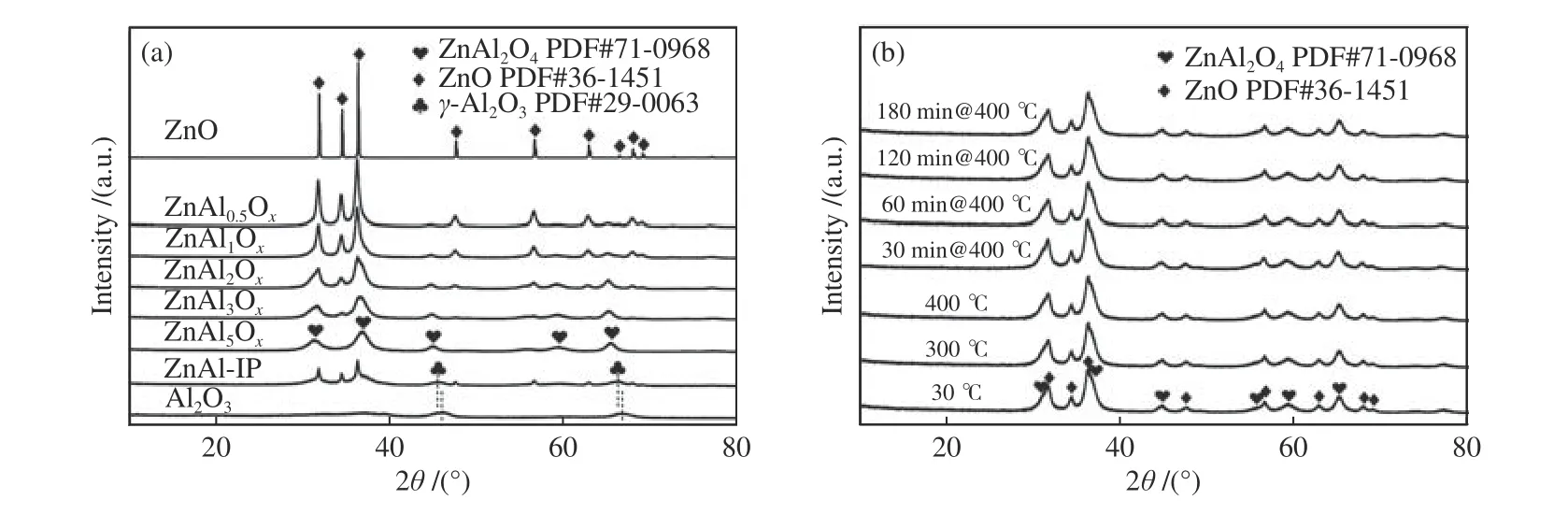
图1 不同Zn-Al 氧化物XRD 谱图及ZnAl2Ox 氧化物的原位XRD 谱图Figure 1 (a) XRD patterns of different Zn-Al oxides; (b) in-situ XRD patterns of ZnAl2Ox oxides treated by H2 at different temperatures and time
图2 为ZnAl2Ox、ZnAl5Ox和ZnAl-IP 氧化物的TEM 和HR-TEM 照片。图2(a)显示ZnAl2Ox氧化物的形貌为明显的豌豆形颗粒且表面光滑,图2(b)中出现的晶格条纹间距为0.243 和0.281 nm,分别归属于ZnAl2O4尖晶石的(311)晶面和ZnO 的(100)晶面[37,38]。在图2(c)中ZnAl5Ox氧化物的豌豆形颗粒形状不明显,在图2(d) 中晶格条纹间距0.243和0.286 nm 分别归属于尖晶石ZnAl2O4的(311)和(220)晶面[39,40],未观察到归属于ZnO 的晶格条纹间距,这与XRD 结果一致。ZnAl-IP 氧化物出现大量纳米颗粒聚集形成的颗粒,颗粒尺寸不均一(图2(e)),图2(f) 中出现了明显的ZnO(002)晶格条纹。
2.2 Zn-Al 样品的织构性质
采用N2吸附-脱附法测定样品的织构性质,如图3 所示。所有样品均呈现IV 型等温线,滞后环为H2 型,说明样品中存在明显的介孔结构。采用浸渍法制备的ZnAl-IP 氧化物的H2a 型滞后环明显减小,这是因为浸渍负载的ZnO 纳米颗粒堵塞了Al2O3的介孔结构。采用M-EISA 法制备的Zn-Al氧化物均为H2b 型滞后环,且随着Zn 含量的增加,滞后环逐渐减小,表明该氧化物中同样存在介孔孔径减小的现象,这是因为过多的ZnO 堵塞了孔道结构,其中,Zn 含量最高的ZnAl0.5Ox中堵塞最为严重。样品的比表面积、孔容和平均孔径列于表1,相比于Al2O3,浸渍Zn 后的ZnAl-IP 比表面积和孔容分别降为111 m2/g 和0.30 cm3/g。采用M-EISA 法制备的Zn-Al 氧化物的比表面积和孔容随着Zn 含量的增加而逐渐降低,平均孔径则出现先增大后降低的趋势。该结果表明,M-EISA 法制备的样品比ZnAl-IP 的孔径和孔容更大。
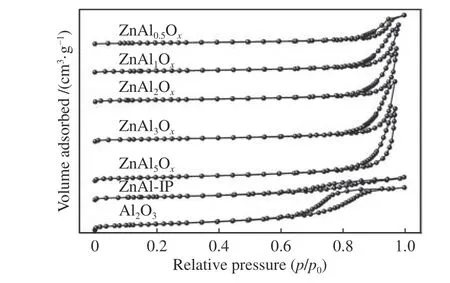
图3 不同Zn-Al 氧化物的N2 吸附-脱附等温线Figure 3 N2 adsorption-desorption isotherms of different Zn-Al oxides
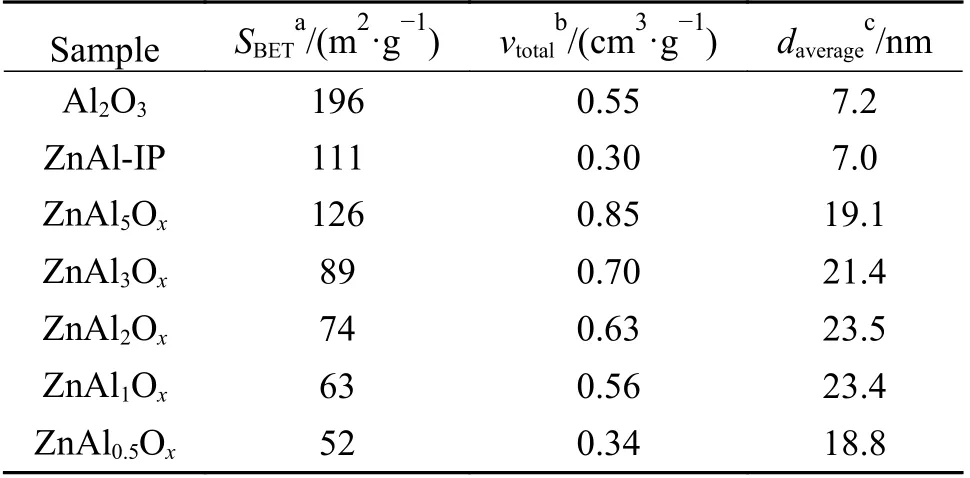
表1 不同Zn-Al 氧化物的织构性质Table 1 Textural properties of different Zn-Al oxides
2.3 Zn-Al 样品的表面性质
采用TPD 表征研究Zn-Al 氧化物表面的化学吸附性质,结果见图4 和表2。图4(a)的CO-TPD曲线显示,单一ZnO 样品仅在710 ℃附近出现微弱的脱附峰,表明ZnO 吸附的CO 量少且不易脱附。ZnAl0.5Ox氧化物的CO 脱附峰为100−800 ℃的宽峰,说明ZnAl0.5Ox中形成的ZnAl2O4尖晶石结构有利于氧化物在中低温区吸附CO。ZnAl2Ox、ZnAl5Ox和ZnAl-IP 氧化物均出现两个明显的脱附峰,分别位于中温区脱附峰(200−400 ℃)和高温区脱附峰(> 400 ℃)。可以明显看出,ZnAl2Ox的中温峰峰强度高于高温峰峰强度,而ZnAl5Ox中高温区脱附峰强弱与其相反。这是因为ZnAl5Ox氧化物中含有相对较多的Al2O3,其脱附峰与单一的Al2O3脱附峰相似[10,41]。ZnAl-IP 氧化物的中高温区脱附峰峰强度相似,这是因为ZnAl-IP 氧化物中的Al2O3载体被表面ZnO 覆盖,减少了高温区CO的吸附。
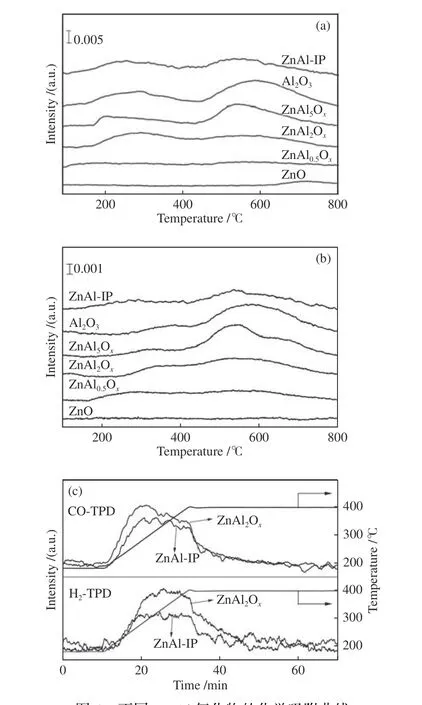
图4 不同Zn-Al 氧化物的化学吸附曲线Figure 4 TPD profiles of different Zn-Al oxides(a): CO-TPD; (b): H2-TPD; (c): CO and H2 TPD profiles in the temperature range of 180−400 ℃ and hold at 400 ℃ for 40 min
图4(b) 的H2-TPD 谱图中ZnO 没有明显的H2脱附峰。M-EISA 制备的Zn-Al 氧化物,随着Al含量的增多,H2脱附峰逐渐向高温区偏移,且总脱附量逐渐增多,见表2,其中,180−400 ℃中温区的H2脱附峰归因于ZnAl2O4尖晶石中−Zn−O−对氢的活化[22,26]。Al2O3样品在400 ℃之前的H2脱附峰较弱,表明Al2O3在中温区对H2的吸附能力弱。ZnAl-IP 氧化物的中温H2脱附峰明显较弱。
为进一步对比ZnAl2Ox和ZnAl-IP 氧化物的中温脱附情况,对该氧化物在180−400 ℃温度进行了程序升温脱附表征,结果见图4(c)。可以看出,ZnAl2Ox比ZnAl-IP 的CO 和H2吸附能力更强,相应的CO 和H2脱附量分别为148.7 和53.1 μmol/g,见表2。该结果表明,M-EISA 制备的ZnAl2Ox比浸渍法制备的ZnAl-IP 具有更多的CO 和H2活性位点。
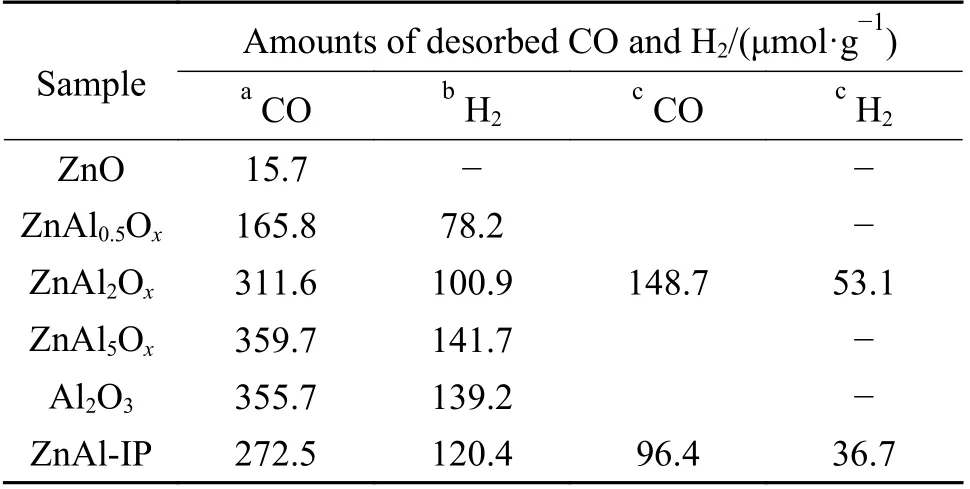
表2 不同Zn-Al 氧化物的CO 和H2 脱附量Table 2 Amounts of desorbed CO and H2 of different Zn-Al oxides
采用H2-TPR 表征金属氧化物的还原性,结果见图5。单一ZnO 和Al2O3样品均未出现明显的还原峰,说明该氧化物难以还原。对于Zn/Al 原子比同为1∶2 的ZnAl-IP 和ZnAl2Ox样品在400 ℃之后出现了还原峰,说明这两个氧化物中Zn 和Al物种状态发生改变,部分氧化物被还原。与ZnAl-IP 样品的还原峰(510 ℃)相比,ZnAl2Ox的还原温度明显往低温(440 ℃)方向偏移,这是因为ZnAl2Ox主要形成ZnAl2O4尖晶石结构,Zn 物种与Al 物种的强相互作用促进了表面ZnO 的还原[28,42],而ZnAl-IP 中主要形成了ZnO 与Al2O3物种,导致ZnAl-IP难以还原。

图5 Zn-Al 氧化物的H2-TPR 谱图Figure 5 H2-TPR profiles of Zn-Al oxides
采用XPS 分析Zn-Al 金属氧化物中各元素的价态和组成,结果见图6。图6(a)为拟合后O 1s的XPS 谱图,图中结合能为529.5−530.6、530.5−531.5、531.5−532.6 eV 三种类型氧的峰,分别对应于晶格氧(OL)、表面氧空位的氧(OV,间接反映表面氧空位含量)以及表面羟基氧的特征峰(OOH)[12,43]。通过拟合后的结果可以看出,ZnAl2Ox的O 1s表面氧空位含量高达40.1%,明显高于ZnAl-IP 氧化物的31.8%。采用氢气气氛(H2/Ar = 2∶18)处理ZnAl2Ox氧化物3 h 后发现,该氧化物表面的氧空位含量进一步增加,达到45.8%。原因可能是ZnAl2Ox氧化物形成的ZnAl2O4尖晶石结构有利于产生氧空位,通过还原性气氛处理后,使氧化物中的部分晶格氧被消耗进而增加了氧空位含量[44]。
为进一步研究Zn-Al 氧化物中的氧空位,采用对氧空位响应灵敏的电子顺磁共振表征方法测试并对比了ZnAl2Ox和ZnAl-IP 氧化物中氧空位的变化,如图6(b)所示。图中g= 2.001 代表氧空位信号[45],可 以 看 出,ZnAl2Ox较ZnAl-IP 峰 信 号 强,表明ZnAl2Ox表面含有更多的氧缺陷位,更有利于反应物CO 的吸附和活化[38]。Liu 等[25]采用EPR测定不同Zn/Al 原子比的样品时也发现,Zn/Al 原子比为1∶2 的氧化物的氧空位浓度最高。
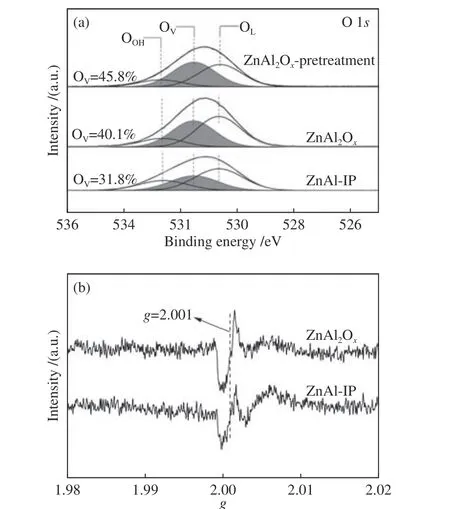
图6 ZnAl2Ox 及ZnAl-IP 氧化物的(a)O 1s XPS 谱图和(b)EPR 谱图Figure 6 (a) O 1s XPS spectra and (b) EPR spectra of ZnAl2Ox and ZnAl-IP oxides
2.4 Zn-Al 样 品 复 合SAPO-18 后 催 化STO 反 应性能
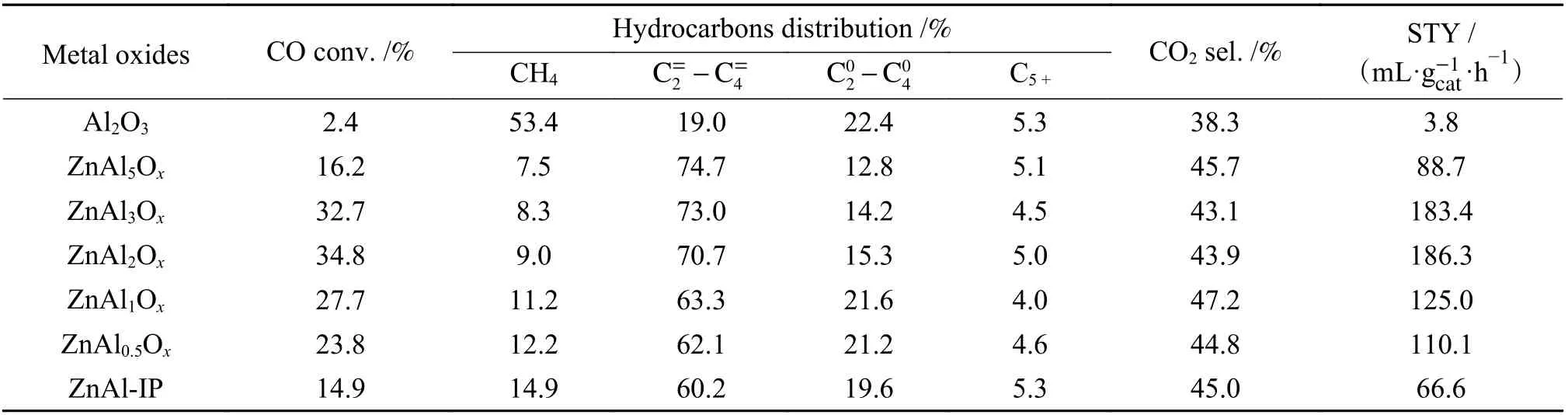
表3 不同Zn-Al 氧化物与SAPO-18 结合后催化STO 的反应性能Table 3 Catalytic performance of different Zn-Al oxides combined with SAPO-18 for STO reaction
图7 为ZnAl2Ox/SAPO-18 和ZnAl-IP/SAPO-18催化剂在反应温度400 ℃、反应压力2.5 MPa、金属氧化物与分子筛质量比为3∶1 时的反应稳定性结果。可以看出,ZnAl2Ox/SAPO-18 催化剂的催化活性和选择性很稳定,且反应50 h 后未出现明显失活。相比于其他文献[25−28]报道的Zn-Al 样品催化STO 反 应,ZnAl2Ox/SAPO-18 的CO 转 化 率 始终维持在34.5% 附近, C=2−C=4选择性始终维持在70.0%附近,这是因为表面具有较多CO 和H2活性位点的ZnAl2Ox氧化物同时具有较大的孔道结构,有利于传质扩散。ZnAl-IP/SAPO-18 催化剂的CO 转化率较低,且 C=2−C=4选择性逐渐下降,烷烃的选择性逐渐上升,这是因为ZnO 纳米颗粒占据了催化剂表面,导致活化CO 的活性位点减少,同时解离的H2相对较多,且ZnAl-IP 的孔径较小,不利于产物扩散,导致反应加氢严重,烯烃选择性下降。
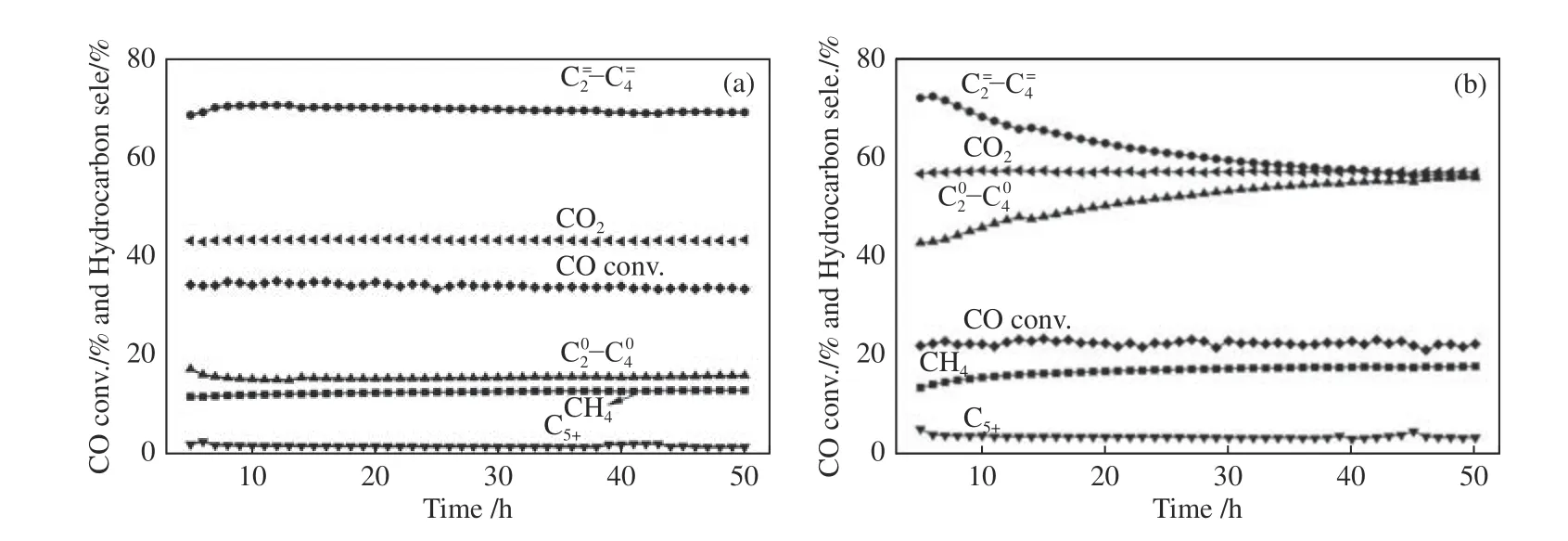
图7 ZnAl2Ox 及ZnAl-IP 与SAPO-18 结合后催化STO 反应的稳定性Figure 7 Catalytic stability of ZnAl2Ox and ZnAl-IP combined with SAPO-18 for STO reaction(a): ZnAl2Ox/SAPO-18; (b): ZnAl-IP/SAPO-18 Reaction condition: 400 ℃, 2.5 MPa, 5000 mL/(gcat·h), weight ratio of Zn-Al to SAPO-18(0.1) is 3∶1,30−60 mesh
3 结 论
通过M-EISA 和浸渍法制备了一系列不同Zn/Al 原子比的Zn-Al 氧化物,并与SAPO-18 物理混合后考察了其催化CO 加氢直接合成低碳烯烃反应性能。当Zn/Al 原子比为1∶5 时,氧化物出现ZnAl2O4尖晶石结构,随着Zn 含量的增加,ZnO 晶相的特征衍射峰逐渐增强。Zn/Al 原子比为1∶2的ZnAl2Ox和ZnAl-IP 氧化物相比,前者具有较大的介孔结构,平均孔径为23.5 nm,CO 和H2吸附量分别达到148.7 和53.1 μmol/g,明显高于ZnAl-IP的吸附量,且其氧空位含量比ZnAl-IP 高8.3%,有利 于 提 高CO 转 化 率。在400 ℃、3.0 MPa 条 件下,M-EISA 法制备的Zn-Al 氧化物随Zn/Al 原子比的增加,CO 转化率呈火山型趋势, C=2−C=4选择性逐渐降低,其中,ZnAl2Ox/SAPO-18 的CO 转化率达到34.8%,时空收率达到186.3 mL/(gcat·h),反应50 h后未出现明显失活。而ZnAl-IP 样品的CO 转化率仅为14.9%, C=2−C=4选择性低且随反应时间的进行明显下降。
猜你喜欢
山东冶金(2022年4期)2022-09-14
耐火材料(2022年4期)2022-08-28
中国宝玉石(2022年2期)2022-04-25
石油炼制与化工(2021年3期)2021-03-23
化工时刊(2020年11期)2020-01-12
中国特种设备安全(2019年9期)2019-12-03
陶瓷学报(2019年5期)2019-01-12
物理学报(2018年5期)2018-03-27
中国有色金属学报(2018年2期)2018-03-26
中国工程咨询(2015年2期)2015-02-14
