
甲酸辅助Cu-ZnO-Al2O3 催化剂制备及其CO2 加氢制甲醇性能研究
2023-02-27 07:35姜秀云杨文兵马清祥高新华赵天生
燃料化学学报 2023年1期
姜秀云 ,杨文兵 ,宋 昊 ,马清祥 ,高新华 ,李 鹏 ,赵天生
(宁夏大学 化学化工学院 省部共建煤炭高效利用与绿色化工国家重点实验室, 宁夏 银川 750021)
化石燃料燃烧释放大量的CO2引发严重的温室效应,导致全球变暖、冰川融化以及极端天气等问题出现,影响人类社会活动,因此,CO2的减排与利用,一直是人们研究的焦点[1,2]。CO2作为一种潜在的碳资源,其碳元素处于最高氧化态,可由H2还原将其转化为大宗基础化学品,如甲醇、甲烷、二甲醚等[3,4]。其中,甲醇可作为基础有机化工原料和液体燃料添加剂,是一种非常有潜在价值的产品[5,6]。将CO2转化为甲醇实现CO2的利用已成为较为活跃的研究领域[7]。
目前,应用于CO2加氢制甲醇反应的催化剂体系主要有铜基催化剂,贵金属催化剂以及其他催化剂。Hu 等[8]将MoS2作为催化剂用于催化CO2加氢制甲醇,通过利用MoS2自身富含的硫空位促进CO2解离,使CO2选择性的加氢合成甲醇,进而提升甲醇的选择性。Han 等[9]以Pt 作为催化剂中的活性位点将其负载于In2O3上,用于CO2加氢制甲醇,结果表明,Pt 可以促进H2的解离,能显著提高In2O3催化剂的甲醇选择性。尽管这两种类型的催化剂对于CO2加氢制甲醇反应具有较好的催化性能,但其对设备要求较高、生产成本大,无法用于工业的大规模生产。相比之下,铜基催化剂催化性能良好、价格低廉,性价比较高。20 世纪60 年代英国ICI 公司首次将Cu-ZnO-Al2O3催化剂应用于工业甲醇合成(合成气制甲醇)[10],由于其对C = O 键具有优异的加氢能力[11],因此,Cu-ZnOAl2O3催化剂也成为CO2加氢制甲醇催化剂的主要研究对象,探究发现该催化剂在催化CO2加氢合成甲醇过程中存在甲醇选择性低、易烧结失活等不足[12,13]。研究者通过添加不同助剂,如Mg[14]、Mn[15]、Fe[16],改变载体,如ZrO2[17]、SiO2[18],以及优化制备方法和条件[19]等来提高催化剂的比表面积、活性位点的分散、表面碱性等,进而提升其催化CO2加氢制甲醇性能。
近年来的研究表明,Cu 基催化剂中Cu+和Cu0均是CO2加氢合成甲醇的活性位点,两者的多少直接影响CO2的转化和甲醇的生成。Wang 等[20]通过蒸氨法合成了Cu/SiO2催化剂,并认为Cu+是活化和转化CO2的活性组分,较高的Cu+/(Cu0+Cu+) 比可提高甲醇选择性。Dong 等[21]采用共沉淀-还原法制备了Cu/Zn/Zr 催化剂,通过改变NaBH4含量控制催化剂中Cu0/Cu+的比,提高催化剂的活性。Shi 等[22]曾采用甲酸辅助固态合成Cu/ZnO 催化剂用于合成气制甲醇,将甲酸用作还原剂与金属硝酸盐发生固相反应形成金属-甲酸盐前驱体(H-COO-M),其在焙烧过程会分解出CO 将Cu2+还原为Cu0,获得的Cu/ZnO 催化剂具有较高活性和甲醇选择性的。
本实验采用甲酸辅助合成Cu-ZnO-Al2O3催化剂用于催化CO2加氢合成甲醇,利用甲酸处理使Cu/Zn/Al 前驱体在惰性气氛的焙烧过程中将Cu2+还原为Cu+和Cu0,改变催化剂中元素间的相互作用,进而提高催化剂的催化性能。该过程省去了传统催化剂的高温H2还原,减少了H2的消耗,具有一定的经济效益。同时本实验探究了不同甲酸量处理对催化剂结构性质、CO2转化率和甲醇选择性的影响。
1 实验部分
1.1 原料与仪器
Cu(NO3)2·3H2O (A.R),上海麦克林生化科技有限公司;Zn(NO3)2·6H2O (A.R)、Al(NO3)3·9H2O (A.R)、无水Na2CO3(A.R)、HCOOH (88%,A.R),国药集团化学试剂有限公司。
微型固定床催化活性评价装置(北京志翔蓝天评价装置技术开发有限公司),反应管内径为8 mm,管长为506 mm;产物分析,GC-9560 气相色谱(上海华爱色谱仪器有限公司)。
1.2 催化剂的制备
采用并流共沉淀法制备质量比为Cu∶ZnO∶Al2O3= 4∶3∶3 催化剂前驱体:按比例称取Cu(NO3)2∙3H2O、Zn(NO3)2∙6H2O、Al(NO3)3∙9H2O配制成1 mol/L的混合金属盐溶液,将其与相同浓度的Na2CO3溶液同时滴加到含有一定量的去离子水的烧杯中剧烈搅拌,控制沉淀温度70 ℃, pH = 7 ± 0.2,沉淀完成后继续搅拌2 h,30 ℃静置老化12 h,过滤、洗涤,80 ℃干燥12 h。
甲酸处理前驱体:将甲酸分别滴加到一定质量的催化剂前驱体中研磨至混合均匀(HCOOH与Cu 物质的量比为0、0.4、0.8、1.2、2.4),120 ℃干燥10 h,N2气氛中以2 ℃/min 的升温速率升至350 ℃焙烧3 h。室温下钝化4 h (钝化气O2/N2(1%/99%)),获得的催化剂命名为xf-CZA (x为HCOOH 与Cu的物质的量比)。所有催化剂样品经过压片、造粒、筛分等过程后,取20−40 目催化剂用于活性评价及表征分析。
1.3 催化剂的表征
采 用 德 国Bruker D8 Advance 型X 射 线 衍 射仪(XRD)对催化剂进行连续扫描,测定催化剂的晶相组成,辐射源为CuKα 射线,管电压40 kV,管电流40 mA,10°–85º扫描。
采用北京精微高博JW-BK122F 型比表面积及孔径分析仪对催化剂比表面积及孔径进行分析。称取0.2 g 催化剂(20−40 目),测定前在300 ℃对样品进行脱气预处理2 h,再通过静态容量法在液氮温度−196 ℃、氮相对压力10−8−10−1测定,比表面积由BET 法计算。
采用美国Micromeritics 公司全自动程序升温化学吸附仪(AutoChem Ⅱ2920) 对催化剂进行程序升温还原(H2-TPR) 表征。称取0.05 g 催化剂(20−40 目) 装在U 型石英管中,升温至150 ℃ (升温速率:10 ℃/min),通入He 吹扫1 h,随后降至室温,切换10% H2/Ar 混合气,再次升温至400 ℃(10 ℃/min),同步在线记录还原曲线。
采用德国Pfeiffer 在线质谱分析分析仪(ThermoStar)对催化剂进行程序升温脱附(CO2-TPD)表征。取0.05 g 催化剂(20–40 目)装在U 型石英管中,通入He 升温至200 ℃ (10 ℃/min),切换H2/CO2/Ar = 70∶23∶7 混合气对催化剂预处理2 h,预处理结束后,温度降至50 ℃。再次通入He 吹扫1 h,切换CO2气体表面反应1 h,最后切换He升温至400 ℃ (10 ℃/min),MS 同步记录CO2脱附曲线。
Cu 比表面积及晶粒尺寸同样由AutoChem II 2920 型化学吸附仪测定。取0.05 g 催化剂 (20–40 目) 装在U 型石英管中,在10% O2/He 混合气气氛围下升温至300 ℃ (10 ℃ /min),对催化剂处理3 h后降至室温,切换10% H2/Ar 混合气,升温至400 ℃(10 ℃/min) 还原1 h,Ar 吹扫降温至50 ℃,换10%N2O/Ar 进行表面反应,结束后用He 吹扫,最后切换10% H2/Ar 混合气,再次升温至400 ℃ (10 ℃/min),同步在线记录变化曲线。反应方程式如下:
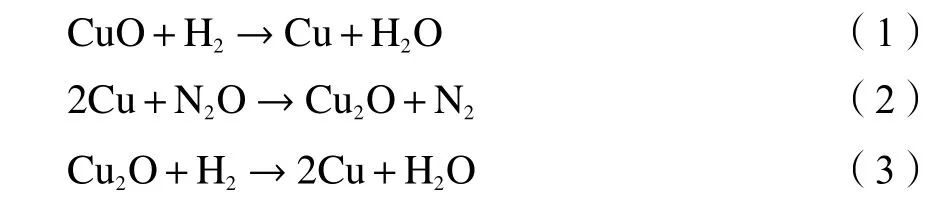
将式(1)中H2的消耗量记为X,式(3)中H2的消耗量记为Y,则可测得:
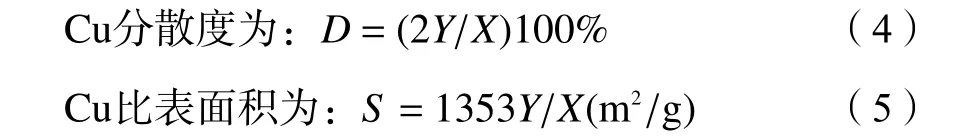
采用法国Setaram 公司的综合热分析仪(SETSYS16)对催化剂进行TG 表征,将催化剂在N2气中由室温升至800 ℃ (10 ℃/min),在线记录曲线。
1.4 催化剂活性评价
催化剂的CO2加氢制甲醇活性测试在微型固定床催化剂评价装置上进行。称取1 g 20−40 目的催化剂样品与2 g 20−40 目的石英砂均匀混合后置于反应管。N2气氛升温至反应温度,切换原料气H2/CO2/Ar = 70∶23∶7 开始催化剂活性评价。评价条件:W/F= 10 g∙h/mol (W为催化剂质量,g;F为原料气流量,mol/h),t= 200 ℃,p= 3.0 MPa, 在线反应时间TOS = 48 h。气相产物经冷凝处理后进入在线气相色谱采样分析,采样时间间隔为2 h。催化剂样品反应活性数据物料平衡以碳为基准计算,使用Ar 作为内标,通过内标法计算CO2转化率和CH3OH 选择性及其产率。计算方法如下:
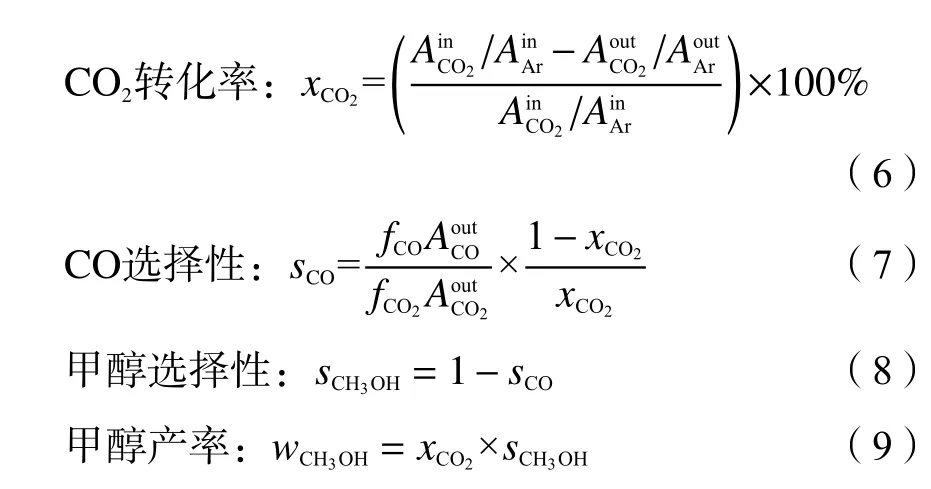
式中,Ai表示组分i的色谱峰面积,μV∙s;fi表示组分i的相对校正因子。
2 结果与讨论
2.1 催化剂的表征
图1 为反应前后催化剂的XRD 谱图。由图1(a)可知,由共沉淀法制备出的前驱体为类水滑石结构Cu3Zn3Al2(OH)6CO3·4H2O (PDF#37-0629)。反 应前的0f-CZA 催化剂只在35.8°处出现弱的CuO 的衍射峰(PDF#45-0937),表明其CuO 的结晶度较低[23,24]。0.4f-CZA 催化剂在2θ为36.4°、42.3°和61.4°出现了Cu2O 的特征衍射峰,分别对应Cu2O 的(111)、(200)、(220)晶面(PDF#05-0667)。0.8f-CZA 催化剂,除了Cu2O 的特征衍射峰强度增强,在2θ为43.3°、50.4°和74.0°处出现了Cu 特征衍射峰,分别对应Cu的(111)、(200)、(220) 晶面(PDF# 04-0836)。对于1.2f-CZA 催化剂,所有衍射峰强度均进一步增强,当HCOOH/Cu 的值再进一步增加到2.4 时,只能观察到Cu 的衍射峰。由此表明随着甲酸量增加,焙烧时释放的还原性气体逐渐增多,使催化剂中Cu 发生连续还原,由Cu2+变为Cu+再继续转变为Cu0。由图1(b)可知,反应后所有催化剂均在2θ=43.3°处出现了明显的Cu 特征衍射峰,这可能是由于反应气本身存在大量的H2,在反应温度下能够将催化剂进行还原。在2θ= 36.4°和32.5°处出现了弱的Cu2O 和CuO 的特征衍射峰,分别对应Cu2O的(111)晶面和CuO 的(110)晶面。所有催化剂在反应前后均未发现任何形式的Zn、Al 氧化物的衍射峰,说明这两者均以无定型和高度分散的形式存在[25]。

图1 反应前后xf-CZA 催化剂的XRD 谱图Figure 1 XRD patterns of xf-CZA catalysts(a): Before reaction; (b): After reaction
图2 为催化剂前驱体的热重分析谱图。由图2(a)可知,0f-CZA 催化剂前驱体的质量损失可归于类水滑石结构各阶段的分解,其中,第 Ⅰ 阶段质量损失发生在50–150 ℃,为物理吸附的水分子蒸发;第 Ⅱ 阶段质量损失发生在150–250 ℃,为类水滑石结构层间水分子的脱除;第 Ⅲ 阶段质量损失发生在250–550 ℃,为类水滑石中羟基基团的脱水和层间碳酸根离子的脱除;第 Ⅳ 阶段质量损失发生在550–650 ℃,为含铜碳酸氧盐的分解,其是在类水滑石结构分解过程中形成[26,27]。甲酸处理的催化剂除了类水滑石结构各阶段的分解,第二阶段处有金属甲酸盐前驱体分解为金属氧化物和CO 等物质的重量损失。由图2(b)的DSC 曲线可知,所有催化剂均在175 和285 ℃左右出现吸热峰,对于0f-CZA 催化剂前驱体归于类水滑石结构的分解,对于甲酸处理的催化剂前驱体则包含类水滑石结构和金属甲酸盐前驱体的分解,此过程中释放大量的CO 和CO2等,而还原性气体CO与催化剂中的Cu2+发生剧烈的氧化还原反应,因此,在210 ℃左右处出现放热峰[22,28,29]。
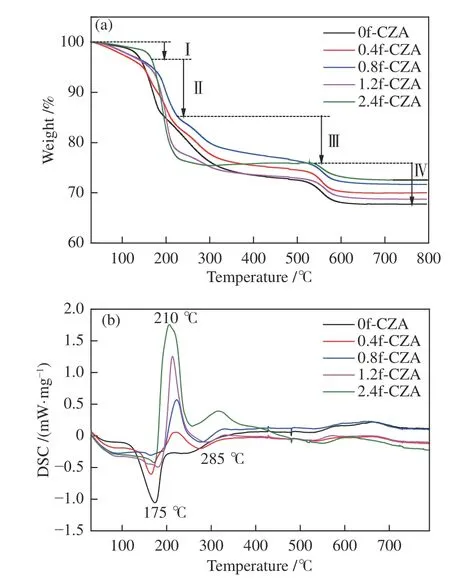
图2 前驱体样品的热分析曲线Figure 2 Thermal analysis curve of precursor samples(a): TG; (b): DSC
图3 为反应前催化剂的吸附-脱附等温曲线和孔径分布。如图3(a)所示,所有催化剂均呈现典型的Ⅳ类等温吸附-脱附曲线。在相对压力较低时,由于过程是可逆的单层吸附,所以曲线相对平稳[30],在相对压力较高时(0.5–1.0)有H3 型回滞环出现,说明催化剂为层状结构聚集的狭缝孔,具有介孔结构。由图3(b)所可知,所有催化剂的孔径均分布在20–50 nm,表明催化剂存在堆积介孔[31,32]。表1 列出了催化剂织构性质参数,由表可知,随着HCOOH/Cu 的值增大,催化剂的比表面积逐渐减小,由N2O 反应吸附法测定的金属Cu 的比表面积(SCu)、Cu 的分散度(DCu)同样呈现减小的趋势。
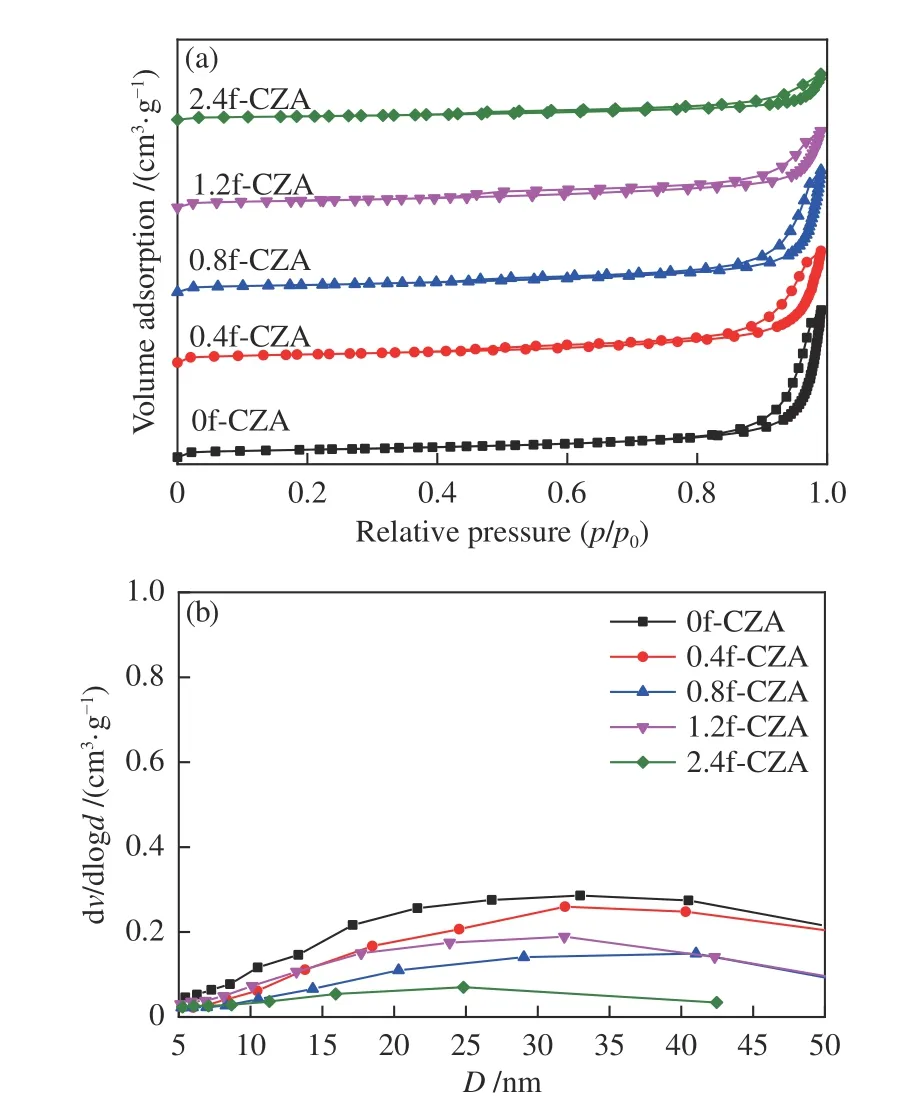
图3 xf-CZA 催化剂的N2 吸附-脱附等温线和孔径分布Figure 3 N2 adsorption-desorption isotherms and pore diameter distribution of xf-CZA catalysts(a): N2 adsorption-desorption isotherm;(b): Pore diameter distribution
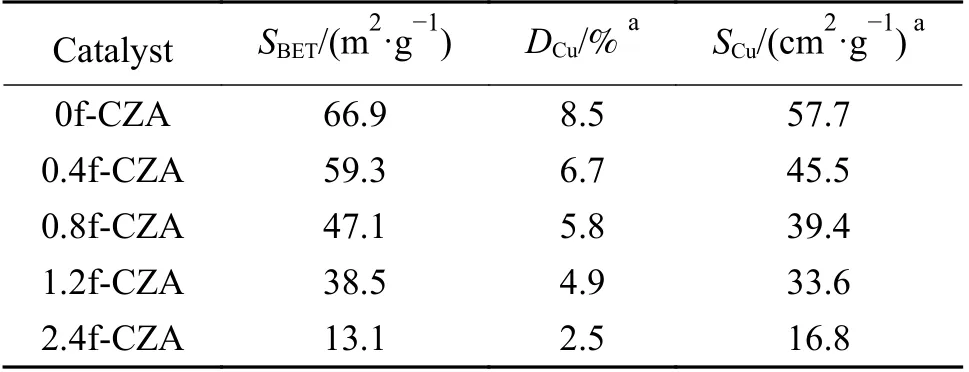
表1 催化剂样品的织构性质参数Table 1 Texture property parameters of catalysts
图4 为反应前催化剂的扫描电镜照片。从图中可以看出,催化剂样品呈现不规则的片状结构分布,对于0.4f-CZA 和0.8f-CZA 催化剂片状结构基本维持,随着甲酸用量增加至2.4f-CZA 催化剂则出现团聚现象,这导致催化剂比表面积减小,与BET 表征结果一致。

图4 xf-CZA 催化剂的SEM 照片Figure 4 SEM images of xf-CZA catalysts(a): 0f-CZA; (b): 0.4f-CZA; (c): 0.8f-CZA; (d): 1.2f-CZA; (e): 2.4f-CZA
图5 为反应前催化剂的H2-TPR 谱图,由于ZnO 和Al2O3的还原温度均高于此温度范围[33],所以图中出现的还原峰均属于催化剂中Cu 物种的还原。对于0f-CZA 催化剂只有一个大的还原峰归于CuO 的还原,将其分为两个高斯峰,其中,在252 ℃左右的还原峰为表面CuO 的还原,307 ℃的还原峰为体相CuO 的还原[34]。对于甲酸处理的催化剂,结合前面XRD 分析可知,这些催化剂中Cu 物种以多种价态混合存在,因此,在不同温度下出现还原峰,较低温度下可归于Cu+还原为Cu0,较高温度下为Cu2+还原为Cu0[35]。观察发现随着甲酸含量的增加较高温度处的还原峰逐渐向低温方向移动,说明甲酸处理改变了Cu 物种与Zn、Al 氧化物间的相互作用,促进了催化剂的还原。还原峰面积逐渐减小,即催化剂的耗氢量逐渐减少,表明催化剂中的部分Cu 在焙烧过程中已经被还原为Cu0,该结论与XRD 表征一致。
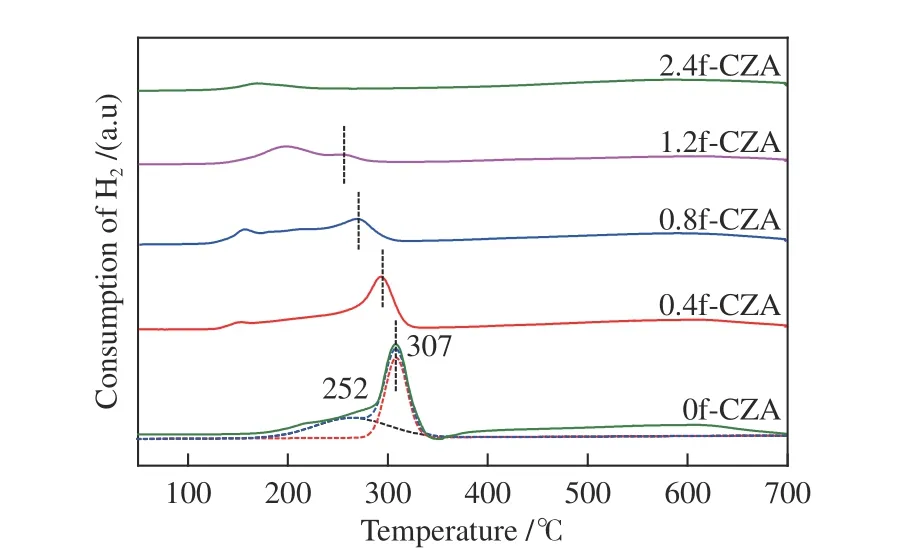
图5 xf-CZA 催化剂的H2-TPR 谱图Figure 5 H2-TPR profiles of xf-CZA catalysts
图6 所示为反应前后催化剂的XPS-AES 谱图,用以分析催化剂表面Cu 物种的化学状态。由图6(a) 可知,0f-CZA 催化剂在结合能为934.5和954.4 eV 左右处出现XPS 峰分别对应于Cu 2p3/2和 Cu 2p1/2,结合940–945 和960–963 eV 左右出现的Cu2+的卫星峰(satellite peak),其是Cu2+离子中价层轨道电子与溢出的光电子相互作用产生的电荷震荡,表明催化剂表面存在Cu2+[36]。经甲酸处理后,催化剂表面的Cu2+峰强度随着甲酸量的增加明显减弱,并逐渐向低结合能方向移动,表明Cu2+逐渐被还原为Cu0或Cu+[37,38],这与反应前催化剂的XRD 以及H2-TRR 表征结果一致。图6(b)中,反应后的催化剂存在微弱的Cu2+的卫星峰,表明反应后的催化剂表面仍有少量的Cu2+存在,可归因于反应过程中有副产物水的存在可能使催化剂表面铜发生氧化,使催化剂表面有少量Cu2+存在,且由于反应后催化剂在表征过程中催化剂与空气接触同样存在部分铜被氧化。图中结合能为952.6 和932.7 eV 左 右 的XPS 峰 对 应 的Cu 2p3/2和 Cu 2p1/2,则可归于Cu0或Cu+。
考虑到Cu0和Cu+结合能的微小差异,XPS 光谱无法区分,因此,测定Cu LMM 俄歇电子能谱以进一步鉴定和量化。如图6(c)和(d)所示,出现肩宽且不对称的Cu LMM XAES 能谱,将其拟合为两个重叠的Cu LMM Auger 动能峰,分配给Cu+和Cu0[39]。表2 中总结了催化剂表面Cu+和Cu0的峰位置以及Cu0/(Cu++Cu0)的比值,由表可知,随着甲酸含量的增加催化剂中Cu0的占比逐渐增大,表明有更多的还原性气体将Cu 物种还原为Cu0。对于反应后的催化剂表面的Cu0/(Cu++Cu0) 的值,显然甲酸处理后的催化剂表面Cu0占比较大,与反应后催化剂的XRD 一致。
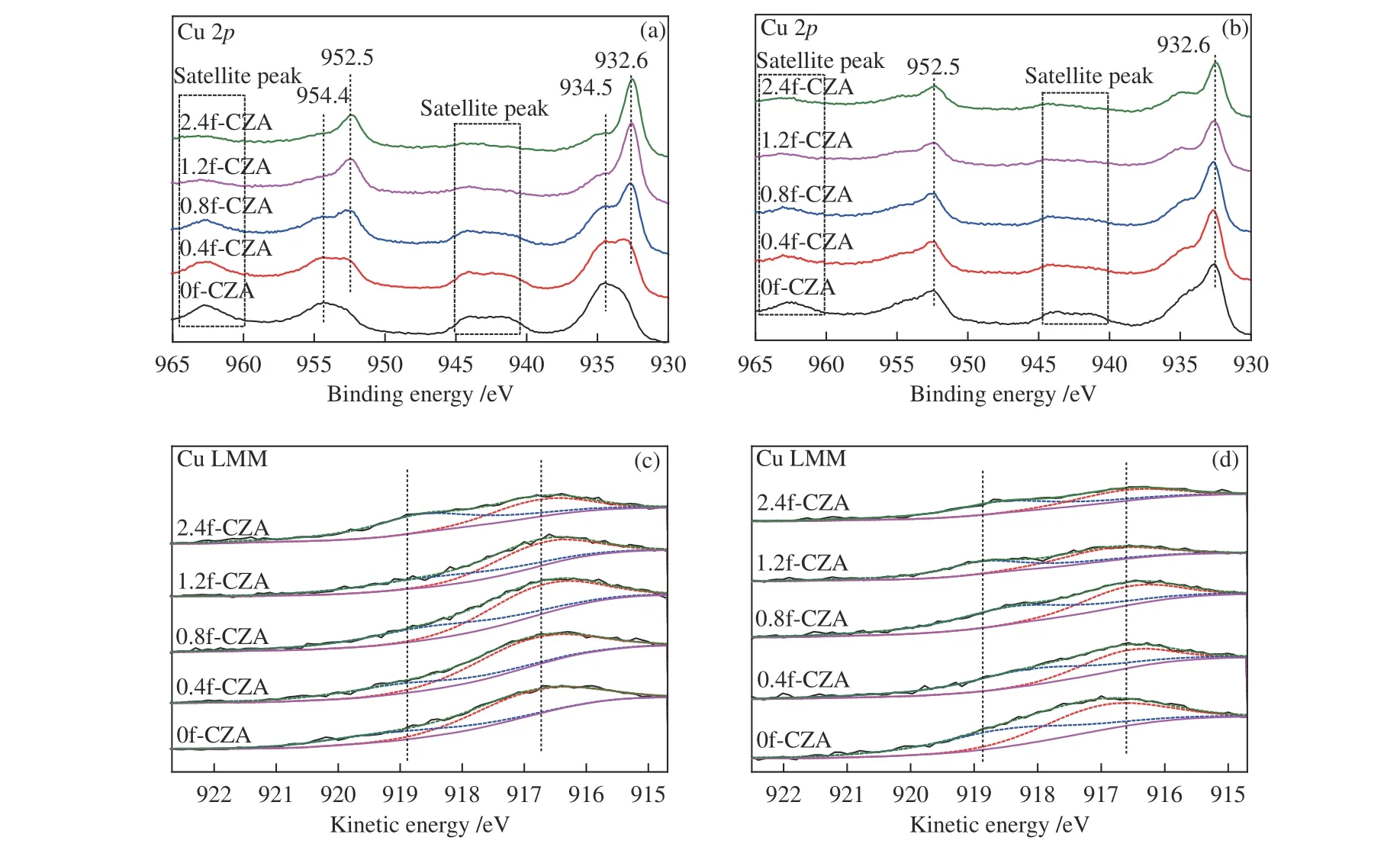
图6 xf-CZA 催化剂的XPS 谱图Figure 6 XPS patterns of xf-CZA catalysts(a): Cu 2p XPS of fresh xf-CZA; (b): Cu 2p XPS of spent xf-CZA; (c): Auger Cu LMM of fresh xf-CZA;(d): Auger Cu LMM of spent xf-CZA
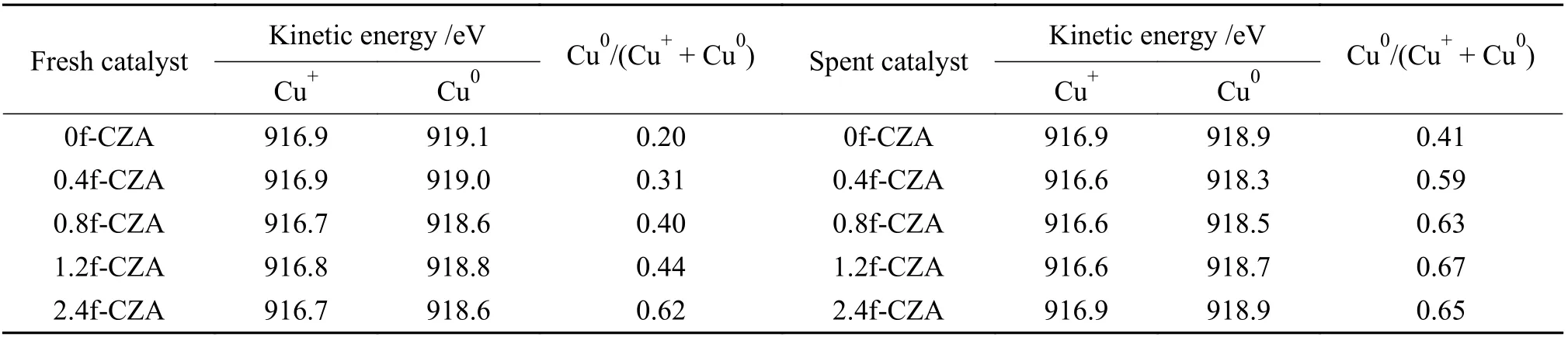
表2 催化剂表面Cu 组分分析Table 2 Copper component analysis on catalyst surface
图7 为反应前催化剂的CO2-TPD 谱图,用于分析催化剂的表面碱性位点,图中的峰面积反映碱量,峰位置反映碱强度,分别对应催化剂的CO2吸附能力以及反应过程中不同中间产物的形成。观察发现,所有催化剂对CO2存在相似的脱附效应,均在50–350 ℃出现了脱附峰,其中,50–150 ℃的CO2脱附峰为α 峰,代表弱碱性位点,150–300 ℃的脱附峰为β 峰,代表中强碱位点。弱碱性位点(α 峰)上的CO2形成的中间体为碳酸氢盐,难于氢化,易于解吸重新转化为CO2。中强碱性位点(β峰)上的CO2形成的双齿碳酸盐中间体会被解离的H 逐步氢化为HCOO*、H2COO*、H2COOH*和H2CO*,最后H2CO*的C = O 键被激活,与表面的H 原子反应形成甲醇[40,41]。与0f-CZA相比甲酸处理后催化剂上的弱碱性位点数减少,中强碱性位点数增加,有助于甲醇的生成。由于催化剂表面的中强碱性与催化剂中金属-氧对(Cu-O、Zn-O、Al-O 等)和表面低配位氧离子O2−有关[42,43],甲酸处理后催化剂在焙烧过程CuO 逐渐被还原为Cu2O和Cu,Cu 与ZnO 界面的接触增加,可能使催化剂表面形成更多的低配位氧氧离子O2−[44],同时元素间的电子效应发生变化促进了金属-氧对的相互作用,从而增加了催化剂表面的中强碱性位点分布[45]。
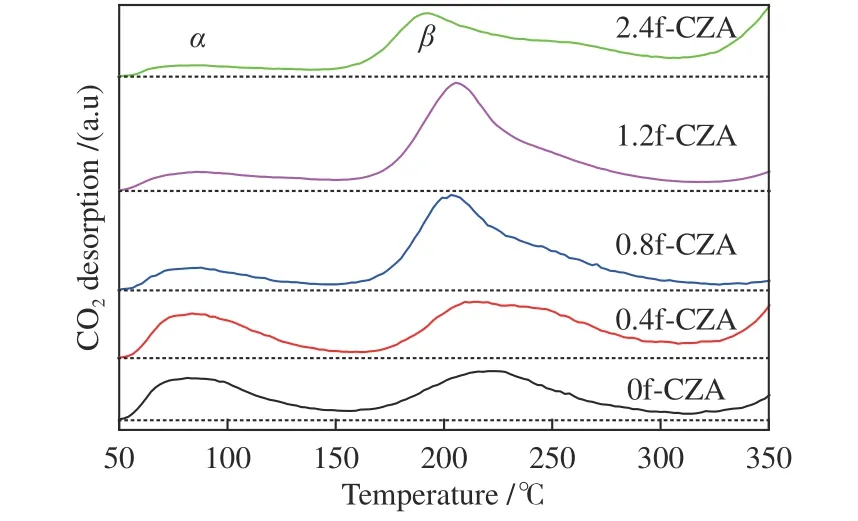
图7 xf-CZA 催化剂的CO2-TPD 谱图Figure 7 CO2-TPD profiles of the xf-CZA catalysts
2.2 催化性能
表3 为催化剂样品的CO2加氢制甲醇活性评价结果表。由表3 可得,甲酸处理对催化剂的活性有明显影响。随着HCOOH/Cu 物质的量比增加,甲醇的选择性明显提高,而CO2转化率略有下降,甲醇产率呈现先增大后减小的趋势。当HCOOH/Cu =0.8 时,CO2的转化率为6.7%,甲醇的选择性达到76.3%,此时甲醇产率最大为5.1%,再进一步增加甲酸的含量时催化剂的活性下降。表明适量的甲酸处理可以改善催化剂的结构性质,提高催化剂的催化活性。
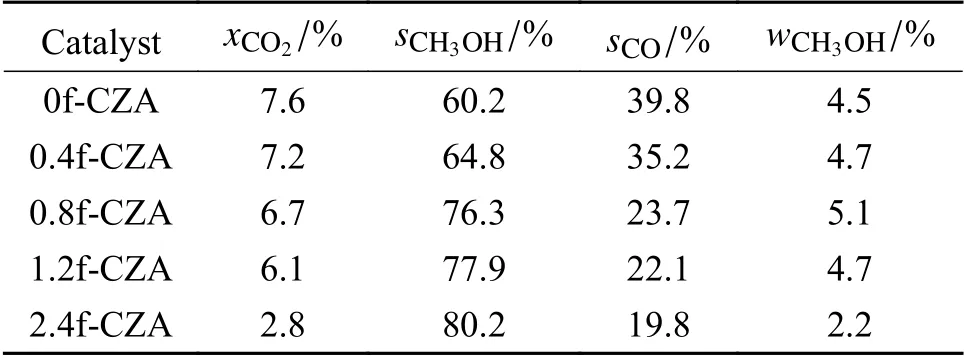
表3 xf-CZA 催化剂活性评价Table 3 Activity evaluation of xf-CZA catalyst
图8 展示了催化剂的催化性能随在线反应时间的变化。由图8 可以看出,所有催化剂在反应20 h 后活性基本稳定,且反应48 h 后转化率和选择性均没有明显的下降趋势,表明催化剂具有良好的稳定性。
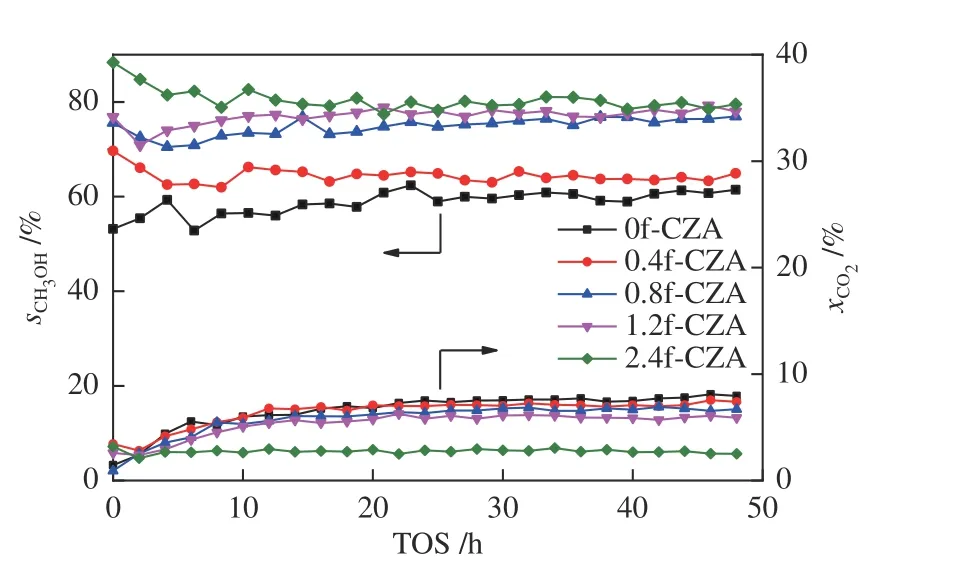
图8 xf-CZA 催化剂的催化性能随反应时间的变化Figure 8 Relationship between the catalytic performance of xf-CZA catalyst and reaction time Reaction conditions: H2/CO2/Ar = 70/23/7, W/F = 10 g·h/mol,p = 3.0 MPa, t = 200 ℃, TOS = 48 h
3 结 论
本实验通过甲酸处理催化剂前驱体制备了系列Cu-ZnO-Al2O3催化剂用于CO2加氢制甲醇反应。研究发现,甲酸处理可以改变催化剂中各元素间的相互作用,使Cu2+物种的还原温度向低温方向移动以及催化剂的中强碱性位点数的增加,有助于催化剂的还原和甲醇的生成。并且甲酸处理可将催化剂中的Cu2+还原为活性中心Cu+和Cu0,可以通过改变HCOOH/Cu 物质的量比调控催化剂中Cu+与Cu0的比例,进一步提升甲醇选择性,促进催化剂活性。200 ℃ 、3.0 MPa 反应条件下,HCOOH/Cu的物质的量比为0.8 时处理获得的催化剂样品具有最高CH3OH 产率(5.1%)。
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
陶瓷学报(2020年2期)2020-10-27
第一财经(2019年8期)2019-08-26
天津医科大学学报(2019年3期)2019-08-13
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中国资源综合利用(2017年4期)2018-01-22
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
