
The Successional Pattern of Microbial Communities and Critical Genes of Consortia Subsisting on Chloramphenicol and Its Metabolites Through Long-Term Domestication
2023-03-22 08:04JiayuZhangKaiyanZhouFangliangGuoHuaxinLeiRenxinZhaoLinLinXiaoyanLiBingLi
Engineering 2023年12期
Jiayu Zhang, Kaiyan Zhou, Fangliang Guo, Huaxin Lei, Renxin Zhao, Lin Lin,Xiaoyan Li, Bing Li,*
a State Environmental Protection Key Laboratory of Microorganism Application and Risk Control, Tsinghua Shenzhen International Graduate School, Tsinghua University,Shenzhen 518055, China
b Shenzhen Engineering Research Laboratory for Sludge and Food Waste Treatment and Resource Recovery, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China
c School of Environment, Tsinghua University, Beijing 100084, China
d Research Center for Eco-Environmental Engineering, Dongguan University of Technology, Dongguan 523808, China
Keywords:Antibiotic Biodegradation Metabolism Microbial community succession Metagenome
ABSTRACT As a widespread emerging contaminant, chloramphenicol (CAP) adversely impacts ecological communities in the water environment.Biological treatment is widely used for aquatic pollutant removal,and the performance of functional microbes determines its outcome.Herein,a consortium with a powerful CAPdegrading capacity was domesticated from activated sludge.As the common degradation products of CAP, 4-nitrobenzoic acid (PNB) and 2,2-dichloroacetic acid (DCA) were also used as the sole substrates for long-term domestication.The successional pattern of the microbial community and critical functional genes through the 2.5-year domestication was revealed by metagenomic analysis.Sphingomonas,Caballeronia, and Cupriavidus became the most dominant populations in the CAP-, PNB-, and DCAdegrading consortia, respectively, and they were crucial degraders of PNB and DCA.Their collaboration contributed to the high mineralization rate of CAP.PNB was transformed into protocatechuic acid(PCA) and then mineralized through meta-cleavage and ortho-cleavage pathways.Crucial functional genes involved in CAP, PNB, and DCA metabolism, including CAP acetyltransferase, CAP oxidoreductase,haloacid dehalogenases, and protocatechuate dioxygenases, were significantly enriched in consortia.pH and carbon source had significant impacts on CAP biodegradation efficiency.The domesticated consortia and isolated strains are necessary microbial resources to enhance the bioremediation of CAP-, PNB-, or DCA-polluted environments.
1.Introduction
Antibiotics play a crucial role in antibacterial treatment in clinics.However,along with excessive consumption,their residues are continuously entering into the environment.Significantly, wastewater from pharmaceutical factories, hospitals, and livestock and aquaculture farms is a main source of antibiotic contamination[1-3].The persistence of antibiotics in aquatic environments negatively impacts ecological communities and facilitates the development and spread of antibiotic resistance genes, which challenges clinical anti-infective therapy [4-6].Removing antibiotics from wastewater can effectively reduce their transfer into the environment, which has become an arduous task in the environmental engineering field [7].Biological treatment systems are commonly applied to remove organic pollutants in wastewater treatment plants (WWTPs).Microbes play a crucial role in removing organic contaminants in engineered environments, and their performance determines the outcomes of biological treatment systems [8].The emergence and prevalence of vigorous antibiotic-degrading microbes will dramatically enhance the removal efficiency of antibiotics in biological treatment systems.The activated sludge microbiome,which is regularly exposed to elevated antibiotic concentrations in wastewater, might serve as a promising resource of antibiotic-degrading microbes that could subsequently be applied in the environmental remediation biotechnology of chloramphenicol (CAP)-contaminated environments.Activated sludge domestication through feeding with the corresponding antibiotic is a conventional approach to remodeling the microbiota with a puissant antibiotic-degrading capacity[9].Researchers have commonly focused on domesticated communities’ functions and microbial structure rather than community succession during domestication[10,11].Determining the processes of microbial community domestication will aid in expanding our knowledge of the successional patterns of microbial communities feeding on antibiotics and provide important information for improving the strategy for microbial community domestication.
As a broad-spectrum antibiotic, CAP is heavily consumed in clinical practice and livestock and aquiculture production in some developing countries owing to its effectiveness and low cost [2].CAP is refractory in wastewater biotreatment systems of WWTPs because of its bacteriostatic activity by inhibiting protein synthesis[12].Herein, we characterized a vigorous CAP-degrading consortium with the potential for application in the enhancement of CAP biotreatment.In our previous study, a comprehensive CAP metabolic pathway was clarified, and 4-nitrobenzoic acid (PNB)and 2,2-dichloroacetic acid (DCA) were proven to be the main CAP biodegradation products produced by this consortium [13].In addition, several studies also pointed out that PNB and DCA were the most common products of CAP in both biological and chemical treatment processes [12,14-16].The residues of PNB and DCA can induce genotoxicity and developmental toxicity in organisms[17].In particular,DCA is also a common noxious disinfection byproduct that generated in the disinfection processes such as chlorination[18].It has already been included in drinking water regulations and guidelines in many countries such as China, the United States,and Canada,because of its adverse effects on human health [19].Thus, it is valuable to domesticate consortia or isolate powerful microbial degraders for efficient DCA and PNB removal from the water environment.The current study was conducted to ①investigate the successional pattern of the CAP-degrading microbial consortium derived from activated sludge during 2.5 years of domestication through metagenomic analysis; ②characterize the CAP, PNB, and DCA degrading capacities of the domesticated consortia and isolated strains; ③decipher the dynamic changes in essential functional enzyme-encoding genes involved in CAP,DCA,and PNB biotransformation;and ④reveal the impacts of environmental and nutrient factors on CAP biodegradation and the microbial community.
2.Materials and methods
2.1.The procedures of consortium domestication
Previously, activated sludge bioreactors treating CAP production wastewater and control bioreactors treating wastewater without CAP were initially established to investigate the development of antibiotic resistance genes in activated sludge under antibiotic selection pressure [20].Herein, they were included in the investigation of the successional pattern of a microbial community subsisting on CAP.A panorama of the entire time course of CAPsubsisting consortium domestication was demonstrated for the first time.The seed activated sludge of these bioreactors was collected from a local municipal WWTP.Detailed information about the operation of these bioreactors has been described in a previous study [20].CAP concentrations in wastewater increased stepwise from 20 to 120 mg·L-1according to their removal behaviors during successive domestication.After approximately 5.5 months, the consortium could steadily degrade 120 mg·L-1CAP in wastewater and was then inoculated into synthetic mineral salt medium(MSM) containing 120 mg·L-1CAP for further domestication to simplify the community.The compositions of MSM were reported in our previous study [13].After approximately 12 months, the consortium was inoculated into MSM containing 100 mg·L-1PNB or DCA for another 12-month domestication to enrich microbes with the ability to utilize PNB and DCA.CAP, PNB, and DCA were supplied as the sole carbon source,and 30 mg·L-1NH4Cl was supplied as a nitrogen source for consortium domestication in MSM.Finally, three consortia named CL-CAP, CL-PNB, and CL-DCA with the ability to utilize CAP, PNB, and DCA as the sole carbon source were successfully domesticated.All cultivation was conducted at 25 °C and 120 revolutions per minute (r·min-1) shaking in batch mode.The culture medium was renewed every 3-5 d according to the consumption of substrates.In this study, 16 microbial samples at each domestication stage were collected for DNA extraction and metagenomic sequencing across the domestication process.In addition,46 metagenomic datasets of consortium CL-CAP reported in our previous studies [20-22] were included for metagenomic analysis with totally different research objectives in this study.Information about all of the involved samples and metagenomic datasets is listed in Table S1.
2.2.Batch biodegradation experiments
Batch biodegradation experiments of CAP,PNB,and DCA by the domesticated CAP-, PNB-, and DCA-degrading consortia were conducted.Cells of the consortia were collected during the batch experiments for metagenomic sequencing.Eight strains were isolated from the domesticated consortium using mineral salt and R2A agar plates amended with CAP[13,21],and they were included for the analysis of their contribution to PNB and DCA biodegradation in the consortium.
To investigate the impacts of various nutrient conditions on the CAP biodegradation performance of the domesticated consortium,MSMs amended with various additional carbon sources(including sodium acetate,sodium citrate,sodium pyruvate,and sodium benzoate at concentrations of 2.4, 12, and 60 mmol·L-1), additional nitrogen sources (including NH4Cl, NaNO2, and NaNO3at 0.56 mmol·L-1), and initial CAP concentration ranging from 50 to 600 mg·L-1were used for batch experiments of CAP biodegradation.Moreover, the impacts of inoculum size (5%, 15%, and 25%of the culture medium volume), pH (3.5, 4.5, 6.0, 7.5, and 8.5),and temperature (20, 25, and 30 °C) on CAP biodegradation by the domesticated consortium were also investigated.Culture cells were collected for 16S ribosomal ribonucleic acid(rRNA)amplicon sequencing at 36 and 72 h after inoculation, except for cultures in MSM amended with sodium benzoate or at pH 3.5 and 8.5 because of their slow growth.Detailed information about these collected samples is listed in Table S2 in Appendix A.
All batch experiments were conducted in triplicate as follows.The cells of domesticated consortium or isolated pure cultures were collected via centrifugation and washed with fresh MSM three times.Then, the collected cells were inoculated into MSM containing the corresponding substrates for batch biodegradation experiments.Liquid samples were collected at intervals for chemical analysis.They were filtered with 0.22 μm polytetrafluoroethylene (PTFE) syringe filters (ANPEL Laboratory Technologies Inc., China).The optical density at 600 nm (OD600) of cultures was determined by an Infinite M200 microplate reader (Tecan,Switzerland).
2.3.Chemical analysis
The soluble total organic carbon (TOC) was determined via a TOC-L analyzer (Shimadzu Corporation, Japan).When CAP, PNB,or DCA was supplied as the sole carbon source in the culture medium, the TOC reduction rate could represent their mineralization rate.The final mineralization rate was calculated as the percentage of TOC reduction.CAP,PNB,PCA,and DCA were detected and quantified by a high-performance liquid chromatography (HPLC)-quadrupole time-of-flight(QTOF)-mass spectrometer(MS)(Impact IITM;Bruker,Germany)equipped with a Thermo Hypersil Gold column (100 mm × 2.1 mm, 1.9 μm) (Thermo Fisher Scientific, USA)according to our previously established method [13].Eluent A of HPLC was Milli-Q water containing 5 mmol·L-1ammonium acetate, and eluent B was methanol.The gradient elution process in HPLC is shown in Table S3 in Appendix A.The MS was operated in negative ionization mode,and the detailed operating parameters for chemical identification and quantification were described in a previous study [13].A reference solution was injected for massto-charge ratio (m/z) calibration.Chemicals were analyzed in scan mode,and the chemical structure was further confirmed according to their fragmentation patterns determined in product ion scan mode.
2.4.DNA extraction,metagenomic,and 16S rRNA amplicon sequencing
Both short-read metagenomes through Illumina sequencing and long-read metagenomes through Oxford nanopore technologies (ONT) sequencing were acquired in this study.The total DNA of microbial samples for Illumina metagenomic sequencing and 16S rRNA amplicon sequencing was extracted using the FastDNATMSpin Kit for Soil (MP Biomedicals, USA) following the manufacturer’s manual.The total DNA of microbial samples for ONT sequencing was extracted using the DNeasy PowerSoil Kit(Qiagen,USA) following the manufacturer’s recommendations.The extracted DNA was checked by 1% agarose gel electrophoresis and a Qubit®2.0 Fluorometer (Life Technologies, USA).Illumina metagenomic sequencing libraries were constructed using the NEBNext®UltraTMDNA Library Prep Kit for Illumina(NEB,USA)following the manufacturer’s instructions.The libraries were sequenced on the Illumina NovaSeq platform using a paired-end(PE) 150 base pairs (bp) strategy at Novogene (China).The construction of the ONT metagenomic sequencing library was performed using the Ligation Sequencing 1D kit (SQK-LSK109) (ONT,UK) following the manufacturer’s recommendations.The ONT library was sequenced on a PromethION (ONT) platform at Novogene (China).Information on 62 short-read metagenomic datasets and ten long-read metagenomic datasets is listed in Table S1.Metagenomic datasets were deposited to PRJNA953649 of the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database.
16S rRNA amplicon sequencing of 104 samples was performed to characterize the microbial community of the consortium under various culture conditions.The V4 region was amplified using primers 515F (5′-GTGCCAGCMGCCGCGG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) on an ABI GeneAmp®9700 polymerase chain reaction (PCR) instrument (Applied Biosystems, USA).The TransStart®FastPfu DNA Polymerase AP221-01 Kit (TransGen Biotech, China) was used for PCR.A 20 μL PCR system consisted of 10 ng of template DNA, 0.8 μL of each primer at 5.0 μmol·L-1,2.0 μL of 2.5 mmol·L-1deoxy-ribonucleoside triphosphates(dNTPs), 4 μL of FastPfu buffer, and 0.4 μL of FastPfu polymerase.The PCR procedures were as follows: initial denaturation at 95 °C for 3 min, 27 cycles of denaturation at 95 °C for 30 s, annealing at 55°C for 30 s,elongation at 72°C for 45 s,and a final extension at 72°C for 10 min.The amplified products were purified using 2%agarose gel and an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, USA).The purified products were quantified using QuantiFluorTMST (Promega, USA).Then, they were sequenced on the Illumina MiSeq platform with a PE 300 bp strategy at Majorbio(China), and the sequencing depth was an average of approximately 50 000 sequences per sample.16S rRNA amplicon datasets were deposited to PRJNA953665 of the NCBI SRA database.
2.5.Metagenomic data analysis
Clean Illumina reads were acquired through quality control using fastp (v0.23.2) [23].The taxonomy of metagenomic data was classified via Kraken2(v2.1.2)[24],and the abundance of each taxonomic level was estimated via Bracken (v2.7) [25].Shannon and Pielou’s evenness indices of the microbial community were calculated using R (v4.1.2).Principal coordinate analysis (PCoA)of samples was performed based on Bray-Curtis distances of communities.
The Illumina reads were assembled using metaSpades(v3.15.4)[26].The ONT long reads were filtered through Filtlong with a minimum length threshold of 500 bp and a minimum mean quality weight threshold of ten.The filtered ONT long reads were assembled using metaFlye (v2.9) [27].The long-read assemblies were polished using Medaka (v1.6.1), Rcon (v1.4.3), and Pilon (v1.24)[28] to obtain high-quality ONT long-read assemblies.Both the short-read and long-read assembled contigs of the final domesticated consortia CL-CAP, CL-PNB, and CL-DCA were assigned to metagenomic assembled genomes (MAGs) via a binning approach using BASALT [29].MAGs were dereplicated by dRep (v2.0.0) [30]with the following parameters: -sa 0.95 -nc 0.30 -comp 50-con 10.The completeness and contamination of MAGs were estimated using CheckM(v1.2.0)[31].The taxonomic classification of MAGs was performed using GTDB-Tk(v2.1.0)[32].The genes in MAGs were predicted by Prodigal (v2.6.3) [33] and annotated by Prokka (v1.14.5) [34] and KofamKOALA [35].Replication rates of MAGs were estimated by CoPTR (v1.1.2) [36].The relative abundances of MAGs in metagenomes were calculated using CoverM(v0.6.1)with the following parameters:genome-m relative_abundance -min-read-aligned-percent 0.75 -min-read-percent-identity 0.95.The significance of the enrichment of each MAG in consortia was determined by the R package limma [37].The relative abundances of selected genes were calculated as follows: First, the aligned read number per base of selected genes was calculated by CoverM with select genes as reference using the following parameters: contig -m reads_per_base -min-read-aligned-percent 0.75 -min-read-percent-identity 0.95.Then, the output was normalized by dividing by the total read number of a dataset to obtain the relative abundance (coverage, ×/Gb) of the selected genes [20].
2.6.16S rRNA amplicon sequencing data analysis
The 16S rRNA amplicon sequencing data were analyzed by the QIIME2 platform [38].The sequences were split and filtered, and then the deblur method was used to denoise and cluster the sequences to generate the operational taxonomic unit(OTU)table.The taxonomy of each representative sequence was annotated by the Ribosomal Database Project (RDP; v2.11) [39] with a confidence threshold of 80%.Redundancy analysis (RDA) and adonis tests were conducted to determine the impacts of environmental and nutrient factors,including the pH,temperature,inoculum size,initial CAP concentration, and initial additional carbon and nitrogen sources, on the microbial community structures using R(v4.1.2).
3.Results and discussion
3.1.Three domesticated consortia with robust CAP, PNB, and DCA degrading ability
The degrading ability of three domesticated consortia for CAP,PNB, and DCA was depicted in Fig.1.Through 2.5 years of domestication with CAP,the final domesticated consortium CL-CAP could completely degrade 250 mg·L-1CAP with a mineralization rate of 93.7%in 36 h(Figs.1(a)and(d)).A comprehensive metabolic pathway of CAP was elucidated in our previous study,and PNB and DCA were found to be the main downstream products of CAP produced by this consortium[13].It has been reported that PNB and DCA are the most common products of CAP in biological treatment processes [12,13,16].In addition, they were also widely detected in the chemical treatment processes of CAP, such as electrochemical degradation and advanced oxidation processes [14,15,40,41].
As a kind of nitrobenzene compound, PNB is an important raw material and mid-product for industrial manufacture.However,its residues in industrial effluents should be eliminated before discharge into the environment because of its genotoxicity and refractory properties[17,42].DCA,a haloacetic acid(HAA),is a common noxious disinfection byproduct with cytotoxic,carcinogenic,genotoxic,teratogenic,and mutagenic properties[18,43].The formation and elimination of HAAs have aroused broad public concern and have been extensively investigated recently [44,45].Herein, two consortia named CL-PNB and CL-DCA were domesticated from CAP-degrading consortia by feeding on PNB and DCA for 12 months, respectively.Consortium CL-PNB completely degraded 250 mg·L-1PNB with a mineralization rate of 95.9% in 14 h (Figs.1(b)and(e)).Consortium CL-DCA completely degraded 100 mg·L-1DCA with a mineralization rate of 91.0% in 8 h (Figs.1(c) and (f)).They demonstrated efficient degradation capacity and have the potential to enhance the bioremediation of PNB- and DCApolluted environments.
3.2.The recovered MAGs and isolated strains of the domesticated consortia
By virtue of the binning approach on both short-read and longread metagenomes, 21 MAGs were recovered from three domesticated consortia(i.e.,CL-CAP,CL-PNB,and CL-DCA)(Fig.2(a)).Three of them(MAG4,MAG5,and MAG6)were complete and consisted of only one circular contig(Figs.2(b) and (c)).13 MAGs were of high quality with completeness higher than 95.0% and contamination less than 3.5% (Fig.2(c)).MAG1, MAG2, MAG3, MAG4, and MAG5 assigned to genera Sphingomonas, Cupriavidus, Caballeronia, Pigmentiphaga,and 62-47 were dominant(average relative abundance>1.0%) in consortium CL-CAP (Fig.2(a)).MAG2 (Cupriavidus),MAG3 (Caballeronia), and MAG4 (Pigmentiphaga) were dominant in consortium CL-PNB, while MAG2 (Cupriavidus) and MAG5 (62-47)were dominant in consortium CL-DCA(Fig.2(a)).MAG1(Sphingomonas), MAG2 (Cupriavidus), and MAG3 (Caballeronia) were significantly(p <0.05)enriched in consortia CL-CAP,CL-DCA,and CLPNB with average relative abundances of 58.6%,92.5%,and 76.5%in the corresponding consortium, implying that they were crucial bacteria responsible for CAP,DCA,and PNB biodegradation,respectively (Fig.2(a) and Fig.3).Replication rates of microbial populations, including MAG1, MAG2, and MAG3, manifested as a reversed-U shaped pattern (Fig.2(a)).This result suggested that MAG1, MAG2, and MAG3 grew and reproduced actively during CAP,DCA,and PNB reduction,which also implied their crucial roles in the biodegradation of these three compounds.
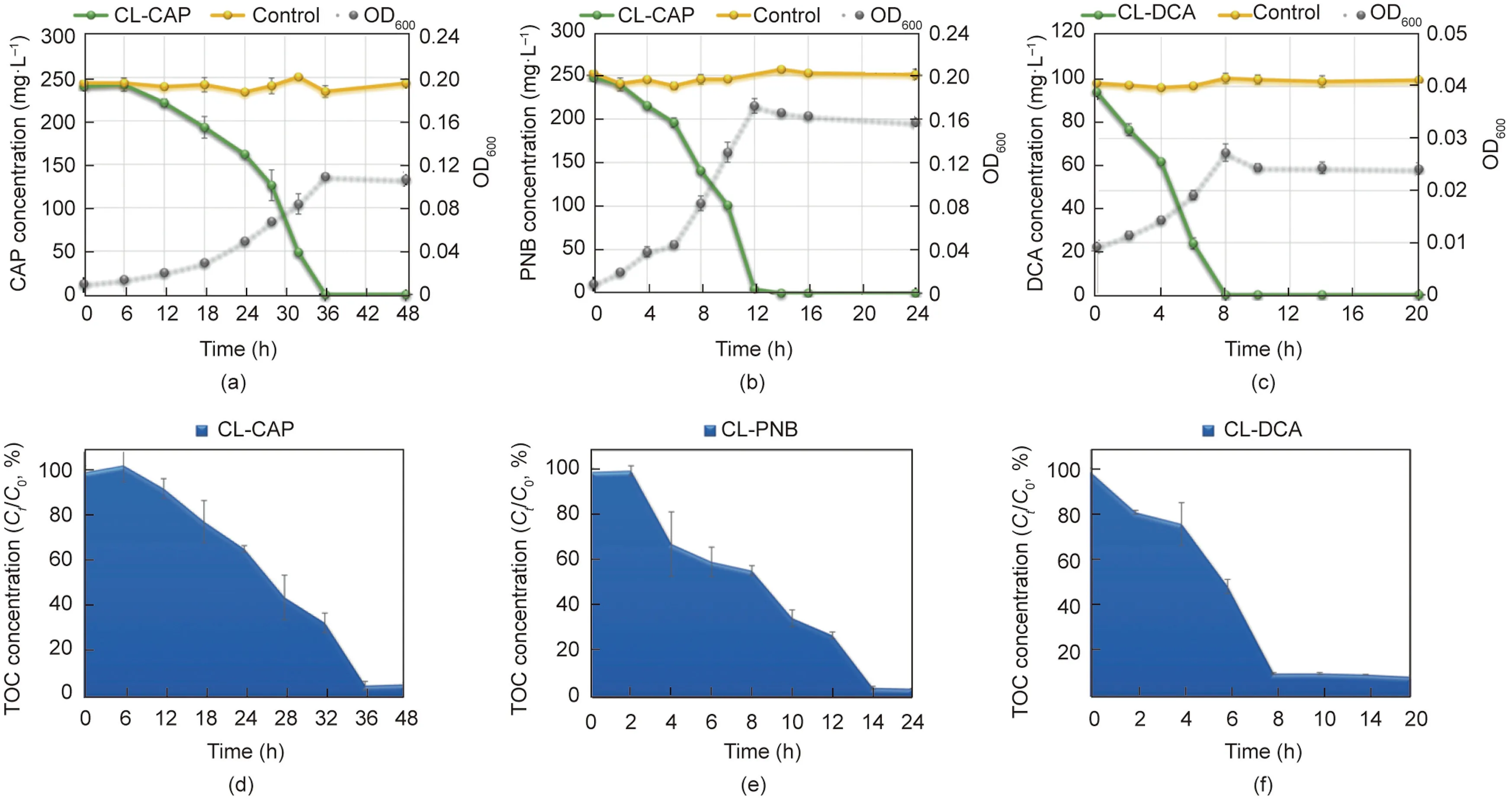
Fig.1.The performance of three consortia on CAP, PNB, and DCA biodegradation after domestication.(a-c): CAP, PNB, and DCA degradation by enriched consortia CL-CAP,CL-PNB,and CL-DCA,respectively.(d-f):CAP,PNB,and DCA mineralization by enriched consortia CL-CAP,CL-PNB,and CL-DCA,respectively.The initial concentrations of CAP,PNB, and DCA were 250, 250, and 100 mg·L-1, respectively.Values are presented as means ± standard deviation (SD) of three replicates.Ct: concentration value at time t;C0: initial concentration value at 0 h.

Fig.2.The features and relative abundances of MAGs in CAP,PNB,and DCA degrading consortia after domestication.(a)The variations of relative abundances and replication rates of MAGs in CAP,PNB,and DCA degrading consortia during the biodegradation process;(b)contig number of each MAG;(c)completeness,contamination,and genome size of each MAG.
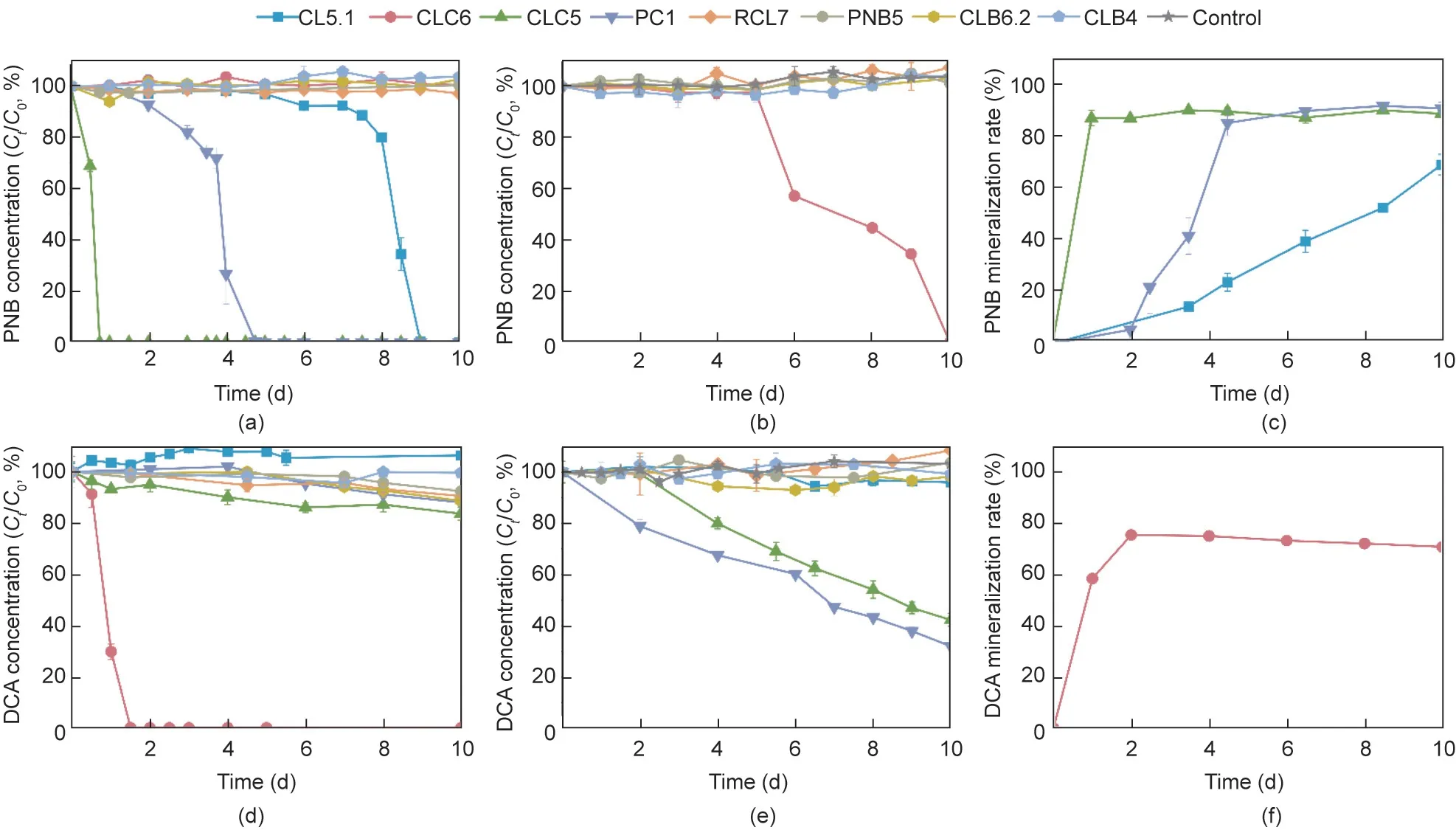
Fig.3.The biodegradation of PNB and DCA by eight isolated strains including Sphingomonas sp.CL5.1, Cupriavidus sp.CLC6, Caballeronia CLC5, Caballeronia PC1,Chryseobacterium sp.RCL7,Labrys sp.PNB5,Pigmentiphaga sp.CLB6.2,and Achromobacter sp.CLB4.(a,d)The biodegradation of PNB and DCA as the sole corban source by eight isolated strains.(b, e) The biodegradation of PNB and DCA in the presence of 500 mg·L-1 sodium pyruvate.(c, f) The mineralization of PNB and DCA by strains that could utilize them as the sole carbon source.The initial concentrations of PNB and DCA were both 100 mg·L-1.Values are presented as means ± SD of three replicates.
Our previous study confirmed that Sphingomonas sp.CL5.1 isolated from consortium CL-CAP was a crucial CAP degrader that could catabolize CAP to PNB and DCA [13].Herein, the PNB- and CAP-degrading capacity of eight strains isolated from consortium CL-CAP was investigated,and the results were shown in Fig.3.Sphingomonas sp.CL5.1,Caballeronia sp.CLC5,and Caballeronia sp.PC1 were PNB degraders who could utilize PNB as the sole carbon source (Fig.3(a)).Cupriavidus sp.CLC6 could also degrade PNB through cometabolism with sodium pyruvate (Fig.3(b)).Among them, Caballeronia sp.CLC5 was the most vigorous PNB degrader,as it could completely degrade 100 mg·L-1PNB in 18 h with a mineralization rate of 89.7% (Figs.3(a) and (c)).Cupriavidus sp.CLC6 was a powerful DCA degrader that could completely degrade 100 mg·L-1DCA in 36 h with a mineralization rate of 70.7%(Figs.3(d)and(f)),while Caballeronia sp.CLC5 and Caballeronia sp.PC1 could also degrade DCA through cometabolism with sodium pyruvate (Fig.3(e)).Thus, we concluded that Caballeronia sp.and Cupriavidus sp.were the main contributors to the catabolism of PNB and DCA converted from CAP by Sphingomonas sp.in consortium CL-CAP.In other words, they collaborated to mineralize CAP in the consortium.These results of pure culture biodegradation tests confirmed the conjecture on the substrate utilizing preferences of predominant members in communities via metagenomic analysis (Fig.2(a) and Fig.S1 in Appendix A).
3.3.The successional pattern of microbial communities subsisting on CAP and its metabolites
The microbial composition across the whole domestication process was revealed by the taxonomy classification of short reads.As shown in Fig.4, 70.22%of the short reads of all samples were successfully classified as bacteria(70.12%),archaea(0.04%),eukaryota(0.05%), and viruses (0.01%).The communities mainly consisted of Proteobacteria (63.57%), Actinobacteria (4.54%), and Bacteroidetes(0.87%) at the phylum level (Fig.4).The microbial diversity and evenness of consortia decreased through domestication, as the alpha diversity indices (Shannon and Pielou’s evenness indices)of the consortia significantly decreased after domestication compared with that of the original seed activated sludge (t test,p <0.05) (Figs.5(a) and (b)).In addition, the community structure was more simplified after domestication in MSM with CAP,PNB,or DCA, as the alpha diversity indices significantly decreased after domestication for more than six months in MSM (t test,p <0.05).Similarly, Luan et al.[46] found that the alpha diversity of the aerobic biofilm reactor markedly decreased because of long-term treatment with streptomycin for 576 d.The PCoA ordination of the communities at the genus level across the domestication process revealed that the microbial community was significantly impacted by the culture medium composition(Adonis test, p <0.001) (Fig.S2(a) in Appendix A).For instance, the microbial community markedly changed after being fed with PNB or DCA(Pairwise Adonis test,p <0.01)(Fig.S2(b)in Appendix A).After laboratory domestication with municipal wastewater for 5.5 months,the control consortium (Control-7) without CAP exposure was mainly composed of Alicycliphilus (19.9%), Acidovorax (10.1%),Diaphorobacter (6.3%), Comamonas (4.5%), and Pseudomonas (3.1%)at the genus level(Fig.5(c)).At the incipient stage of domestication with 20 mg·L-1CAP, Paenarthrobacter (46.0%), Alicycliphilus (4.4%),and Microbacterium (4.2%) prevailed in the community (CAP-20)(Fig.5(d)).After domestication with stepwise increasing CAP concentrations for 5.5 months,Paenarthrobacter(13.9%),Sphingomonas(12.9%), and Caballeronia (6.8%) became predominant in the community (CAP-120) (Fig.5(d)).Finally, through another two-year domestication in the MSM, Sphingomonas (57.8% in CAP-120c),Caballeronia (60.2% in PNB-100), and Cupriavidus (74.5% in DCA-100) became dominators in the communities subsisting on CAP,PNB, and DCA, respectively (Fig.5(d)).Sphingomonas, Caballeronia,and Cupriavidus were the key degraders of CAP, PNB, and DCA,respectively, which was proven by biodegradation tests using the corresponding pure cultures in this study and our previous study[13].They became prevalent by virtue of their advantage of substrate utilization in communities feeding on CAP, PNB, and DCA,respectively.
3.4.Dynamic changes in critical genes involved in CAP, DCA, and PNB biotransformation

Fig.4.The taxonomical structure from kingdom to order levels of the microbial communities throughout the whole domestication stage.The thickness of a link indicates the average relative abundance of a taxonomy.
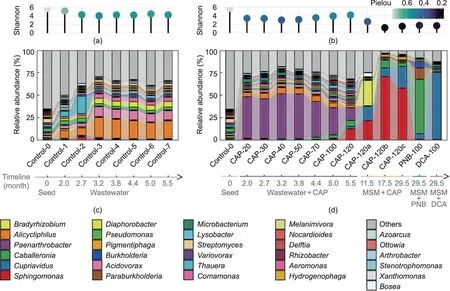
Fig.5.The microbial succession of the communities during the 2.5-year domestication.(a)Shannon and Pielou’s evenness indices of the control consortium communities;(b)Shannon and Pielou’s evenness indices of the consortium communities enriched by CAP,PNB,and DCA,respectively;(c)average relative abundances of dominant genera in the control consortium communities; (d) average relative abundances of dominant genera in the consortium communities enriched by CAP, PNB, and DCA, respectively.Values are presented as means of samples in each group.
Our previous study found that CAP was mainly degraded via acetylation and oxidization by the consortium CL-CAP[13].Acetylation is a common CAP-resistant pathway for bacteria because the biodegradation product CAP 3-acetate loses antibacterial activity[47].The gene catB3 encoding CAP acetyltransferase, which is responsible for CAP acetylation, was discovered in MAG2 (Cupriavidus).The average relative abundance of catB3 was 3.7 in the control consortium without CAP exposure as shown in Fig.6.When domesticated with wastewater and then synthetic MSM containing CAP,the average relative abundance of catB3 increased from 4.0 to 62.4.The oxidization of the hydroxyl group to the carboxyl group at C3of CAP was a CAP biodegradation pathway reported recently by our team [13,48] and Ma et al.[12].The oxidoreductase capO belonging to the glucose-methanol-choline(GMC)oxidoreductase superfamily was discovered to catalyze the two-step oxidation of CAP into oxidized CAP in Sphingomonas sp.CL5.1 in previous studies [48,49].The relative abundance of capO was less than 0.05 in the control consortium without CAP exposure.When domesticated with wastewater and then synthetic MSM containing CAP,the average relative abundance of capO increased from 25.4 to 341.6.
Haloacid dehalogenases, which can catalyze the hydrolysis of the carbon-chlorine bond in DCA, widely exist in aerobic bacteria[50,51].This study discovered 15 gene sequences belonging to four types of haloacid dehalogenase-encoding genes (including dhlB,dehI, dehII, and hdl IVa) in community members, including MAG2(Cupriavidus), MAG3 (Caballeronia), MAG5 (62-47), MAG8 (Bosea),MAG10 (Afipia), MAG13 (Achromobacter), and MAG15 (Mesorhizobium).There were four haloacid dehalogenase-encoding genes in the genome of MAG2 (Cupriavidus), and they were at higher relative abundances in consortium CL-DCA than in other consortia(t test, p <0.05).In fact, the isolated strain Cupriavidus sp.CLC6 indeed exhibited a strong DCA-degrading ability.
PCA was detected to be a product of PNB produced by consortium CL-PNB,and its structure was further confirmed by PCA standards using HPLC-QTOF-MS (Figs.S3(a) and (b) in Appendix A).PCA was produced during PNB biodegradation and finally mineralized by consortium CL-PNB (Fig.S3(c) in Appendix A).Herein, the PCA-degrading capacity of isolated PNB degraders was also tested.The isolated PNB degraders, including Sphingomonas sp.CL5.1,Caballeronia sp.PC1, and Caballeronia sp.CLC5 could utilize PCA as the sole carbon source (Fig.S3(d) in Appendix A).In particular,Caballeronia sp.PC1 and Caballeronia sp.CLC5 completely degraded 10 mg·L-1PCA in 8 h.These results suggested that PNB was transformed into PCA and then catabolized via the PCA cleavage pathway by the domesticated communities.PCA is one of the most common intermediates in the metabolic pathways of various aromatics [52].Usually, PCA can be further catabolized via 4,5-cleavage (meta-cleavage), 3,4-cleavage (ortho-cleavage), and 2,3-cleavage pathways [52,53] by aerobic bacteria.We inferred that the domesticated communities mainly catabolized PCA via metacleavage and ortho-cleavage,as the corresponding critical catalyzing enzymes were identified in MAGs.Protocatechuate 4,5-dioxygenases composed of an alpha chain (ligA) and a beta chain(ligB) are responsible for catalyzing the meta-cleavage of PCA into 4-carboxy-2-hydroxy-cis,cis-muconate 6-semialdehyde [54].The corresponding genes ligA and ligB were discovered in MAG1(Sphingomonas), MAG3 (Caballeronia), and MAG4 (Pigmentiphaga), indicating their potential in meta-cleavage of PCA (Fig.6).The orthocleavage of PCA is catalyzed by the protocatechuate 3,4-dioxygenase, which consists of two subunits (protocatechuate 3,4-dioxygenase alpha chain (pcaG) and protocatechuate 3,4-dioxygenase beta chain (pcaH)) [55].Ten MAGs were identified to harbor both pcaG and pcaH with a potential capacity for PCA ortho-cleavage (Fig.6).As shown in Fig.6, the identified gene sequences of pcaG and pcaH formed three main clades in the phylogenetic tree.Genes pcaG and pcaH carried by MAG2(Cupriavidus)and MAG3(Caballeronia)were significantly enriched in the domesticated consortia (CAP-120c, PNB-100, and DCA-100) (t test,p <0.05).These results demonstrated the dynamic changes in the relative abundances of the critical genes involved in CAP,DCA,and PNB biotransformation during long-term domestication.
3.5.Impacts of environmental and nutrient factors on the CAPdegrading microbial community
Determining the factors influencing the performance of the CAP-degrading consortium is beneficial in guiding the design of the strategy for its application in the enhancement of CAPcontaminated environment bioremediation.Herein, the performance of CAP degradation by the domesticated CAP-degrading consortium CL-CAP under various environmental and nutrient conditions was investigate,and the results of CAP biodegradation and biomass of the consortium were shown in Figs.S4 and S5 in Appendix A.Moreover,the effects of the tested environmental and nutrient conditions on the populations of consortium CL-CAP were determined by 16S rRNA amplicon sequencing.As shown in Fig.S4(a), consortium CL-CAP degraded CAP faster as the initial inoculum size increased from 5% to 25%.It could completely degrade 240 mg·L-1CAP in 28 h at an initial inoculum size of 25%,which was more efficient than the known CAP degraders[12,16,56].Consortium CL-CAP could completely degrade 240 mg·L-1CAP at temperatures ranging from 20 to 30 °C, and it was fastest at 30 °C (Fig.S4(b)).The CAP-degrading efficiency of consortium CL-CAP was highest at a pH value of approximately 6.0,and the CAP degradation rate and cell growth were observably inhibited at pH values of 8.5 and 3.5 (t test, p <0.01) (Figs.S4(c)and S5(c)).CAP at initial concentrations ranging from 50 to 400 mg·L-1could be degraded entirely by the consortium, while CAP at initial concentrations higher than 500 m·L-1was hard to degrade (Fig.S4(d)).CAP exerts antimicrobial effects by binding to the ribosomal 50S subunit, thereby impeding protein synthesis in bacteria [57].When the CAP concentration is too high for community populations to detoxify in time,the growth of the community populations will be inhibited (Fig.S5(d)), and thereby, their substrate utilization rate will slow down.
Consortium CL-CAP was able to utilize CAP as the sole carbon and nitrogen source(Figs.S4(e)and(f)).The presence of additional carbon sources did not markedly accelerate the biodegradation of CAP by the consortium.CAP biodegradation was even hampered by sodium acetate, sodium citrate, and sodium benzoate (Fig.S4(e)).Remarkably, sodium acetate and sodium benzoate at concentrations higher than 12 mmol·L-1significantly inhibited CAP biodegradation (t test, p <0.01; Fig.S4(e)).Our previous study found that the growth of Sphingomonas sp.CL5.1, the critical CAP degrader in consortium CL-CAP, was significantly inhibited by 6 mmol·L-1sodium acetate [13], which probably was a crucial obstructive factor for CAP biodegradation by the consortium.In addition, the biomass of community populations did not increase in the presence of 12 and 60 mmol·L-1sodium benzoate (t test,p < 0.01; Fig.S5(e)).Sodium benzoate exerts broad-spectrum antimicrobial activity by interfering with the permeability of microbial cell membranes and inhibiting the activity of the respiratory enzyme system[58].Thus,it is widely used in many foods and soft drinks as a preservative and antimicrobial agent [59].Herein,the growth of the CAP-degrading consortium was intensely inhibited by sodium benzoate,which might serve as an approach for the control and disinfection of this consortium once it has completed its job for CAP removal.The inhibitory effect of nitrite nitrogen on CAP biodegradation and growth of the consortium was also observed (Figs.S4(f) and S5(f)).Several studies have reported that nitrite nitrogen has an inhibitory effect on bacterial growth and contaminant removal in biological wastewater treatment [60,61].Nitrite nitrogen was proven to interfere with the cellular respiration of aerobic microbes by deactivating cytochrome oxidases or iron-containing enzymes [62].
The RDA suggested that the community was strongly affected by pH, initial CAP concentrations, and additional carbon sources(Adonis test,p <0.05)(Figs.7(a)and(b)).Pigmentiphaga and Labrys exhibited a positive relationship with pH (Fig.7(a)).The relative abundance of Labrys was less than 0.5% at pH values of 4.5 and 6.0,while it was higher than 1.7%at pH values of 7.5 and 8.5.Moreover,Labrys even became a dominant member with an average relative abundance of 27.2% in the consortium at pH 8.5 (Fig.S6 in Appendix A).Cupriavidus showed a positive relationship with the initial CAP concentrations (Fig.7(a) and Fig.S6).The community populations were intensely affected by additional carbon sources,including sodium acetate, sodium citrate, sodium pyruvate, and sodium benzoate,because of their dissimilar substrate preferences(Fig.7(b) and Fig.S6).The demonstration of the impacts of environmental and nutrient factors on the CAP-degrading microbial community could guide the improvement of the biotreatment of CAP-polluted wastewater for better outcomes.
4.Conclusions
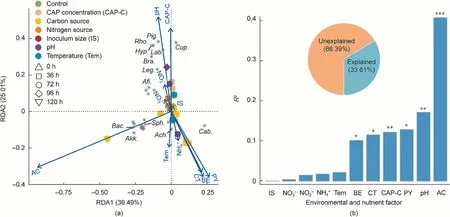
Fig.7.The impacts of environmental and nutrient factors on the CAP-degrading microbial community.(a) Impacts of environmental and nutrient factors on the microbial community structures at genus level revealed by the RDA.(b) Adonis analysis determining the contribution of each environmental and nutrient factor on the microbial community structure.The pie shows the percentages of explained and unexplained variations.The tested carbon sources included sodium acetate(AC),sodium pyruvate(PY),sodium citrate (CT), and sodium benzoate (BE).The tested nitrogen sources included ammonium chloride (NH4+), sodium nitrate (NO3-), and sodium nitrite (NO2-).Sph.:Sphingomonas; Cup.: Cupriavidus; Cab.: Caballeronia; Pig.: Pigmentiphaga; Lab.: Labrys; Bra.: Bradyrhizobium; Ach.: Achromobacter; Hyp.: Hyphomicrobium; Rho.: Rhodopseudomonas; Lei.: Leifsonia; Afi.: Afipia; Leg.: Legionella; Akk.: Akkermansia; Bac.: Bacteroides; *p <0.05, **p <0.01, and ***p <0.001.
Biological treatment is a conventional and economical approach for antibiotic elimination in the practice of environmental bioremediation, such as wastewater treatment.The outcome is determined by the performance of functional microbes in the system.The performance of a biological treatment system can be enhanced via domestication with the corresponding antibiotic.Herein, three consortia with powerful CAP-, PNB- and DCA-degrading capacity were successfully domesticated from activated sludge.The successional pattern of microbial communities and critical genes of consortia through the 2.5-year domestication was characterized.Finally,Sphingomonas(57.8%),Caballeronia(60.2%),and Cupriavidus(74.5%)became dominators in the communities subsisting on CAP,PNB, and DCA in the MSM, respectively.Overall, the microbial diversity and evenness of consortia decreased through domestication.Crucial genes involved in CAP, PNB, and DCA metabolism were significantly enriched in consortia through long-term domestication.Environmental and nutrient factors such as pH and carbon source had observable impacts on the degradation efficiency,which should be considered for its further application in environmental bioremediation.Sphingomonas, Caballeronia, and Cupriavidus were the key degraders for CAP, PNB, and DCA in these consortia,respectively,due to their advantage of substrate utilization in communities.The domesticated consortia and isolated strains showed excellent performance on CAP, PNB, and DCA mineralization, which are important microbial resources for bioremediation.It should be noted that the stable colonizing capacity of the domesticated consortia and pure cultures in the practice of bioremediation should be further studied.
Acknowledgments
This study was funded by National Key Research and Development Program of China (2022YFE0103200), National Natural Science Foundation of China (22176107 and 22206107),Guangdong Basic and Applied Basic Research Foundation(2019B151502034 and 2021A1515110772), and China Postdoctoral Science Foundation (2021M691772).
Compliance with ethics guidelines
Jiayu Zhang, Kaiyan Zhou, Fangliang Guo, Huaxin Lei, Renxin Zhao,Lin Lin,Xiaoyan Li,and Bing Li declare that they have no conflict of interest or financial conflicts to disclose.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2023.07.009.

- Engineering的其它文章
- Global Top Ten Engineering Achivements 2023
- 2023 Global Engineering Fronts
- Will Massive Appetite for Minerals Stall Clean Energy Transition?
- Optical Microscopy Advances Reach Sub-Nanometer Resolution
- International Correlation Research Program: Cross-Fault Measurement for Earthquake Prediction
- A Systematic Perspective on Communication Innovations Toward 6G