
基因组分析对猪乳头数相关数量性状基因座鉴定
2023-05-17 06:57尹彦镇侯黎明刘航陶伟石传宗刘锴月张萍牛培培李强李平华黄瑞华
中国农业科学 2023年10期
尹彦镇,侯黎明,刘航,陶伟,石传宗,刘锴月,张萍,牛培培,李强,李平华,黄瑞华
基因组分析对猪乳头数相关数量性状基因座鉴定

1南京农业大学养猪研究所,南京 210095;2南京农业大学淮安研究院,江苏淮安 223001;3淮安市淮阴新淮种猪场,江苏淮安 223322
【目的】分析乳头数的变异,挖掘与乳头数相关的数量性状基因座(quantitative trait locus,QTL)和候选基因,为猪乳头数的选育研究提供重要分子标记。【方法】准确测定了709头苏淮猪(335头育肥猪和374头种猪)的左、右和总乳头数。对苏淮育肥猪进行80K芯片分型,并使用芯片数据计算左、右、总乳头数的遗传力和基因组估计育种值(genomic estimated breeding value, GEBV)。基于乳头数GEBV和表型排名,选择前10%的个体以及后10%的个体进行群体分化指数分析(fixation Index,F)检测高度分化的位点。接着,通过全基因组关联分析(genome wide association analysis, GWAS)鉴定与乳头数关联的位点,选择高度分化且与乳头数显著关联的位点作为候选位点,选择位于候选位点附近且功能注释后与乳头数相关的基因作为候选基因。最后,选择每个染色体上最显著的候选位点对709头苏淮猪进行乳头数关联分析,以验证上述位点的显著性。【结果】苏淮育肥猪左、右、总乳头数的变异系数分别为10.20%、9.26%、8.50%,遗传力分别为0.212、0.257、0.312。基于F和GWAS分析,总共在7、13、16、18号染色体(Chromosome, SSC)上鉴定到20个乳头数的候选位点,这些候选位点可解释5.49%—8.03%的表型方差。其中,SSC7上与总乳头数关联的位点rs80894106与文献中报道的影响大白和杜洛克猪总乳头数的候选位点一致,但左乳头的候选位点rs81444134(26.51 Mb, SSC13)和rs81233299(8.13 Mb, SSC18)均为新发现的与乳头数相关的位点。左、右、总乳头的候选位点主要集中在SSC16上的6.36—10.66 Mb区间;连锁不平衡(linkage disequilibrium, LD)分析发现,区间内7.47—8.27 Mb的候选位点拟合成了一个795 kb的单倍型块,且该单倍型块是新发现的影响乳头数的候选区域;单倍型块内的rs337606862(7.47 Mb)与右乳头和总乳头最显著关联,单倍型块内的3个位点均位于cadherin 18()基因的内含子上,编码Ⅱ型钙黏附素,且钙黏附素与发育中组织细胞的识别、分选、增殖、凋亡以及乳腺癌的发生有关。因此,可能是新的影响猪乳头数的候选基因。再者,本研究对4个染色体上最显著的位点:rs81444134、rs80894106、rs337606862、rs81233299在709头苏淮猪中基因分型,经关联分析后发现,这些位点均与乳头数显著相关,可以作为潜在分子标记用于乳头数的选育。【结论】本研究通过基因组分析在苏淮猪群体中鉴定到20个与乳头数显著相关的位点。其中SSC13上的26.51 Mb和SSC18上的8.13 Mb是新的乳头数的候选QTLs,SSC16上的7.47—8.27 Mb也是新发现的乳头数的候选QTL,且区间内可能是新的影响猪乳头形成的候选基因。
F;GWAS;数量性状基因座(quantitative trait locus,QTL);苏淮猪;乳头数;候选基因
0 引言
【研究意义】猪的乳头一般呈圆柱状,分布在腹部中线的两侧。根据分布位置的差异,猪的乳头划分为左乳头和右乳头。发育正常的乳头具备泌乳的功能,能够为仔猪提供生长发育所必须的营养物质。因此,拥有更多功能正常乳头的母猪能够哺乳更多的初生仔猪。研究表明,仔猪利用的母猪功能乳头的数目与仔猪的成活率密切相关[1]。同时,KIM等[2]研究了商业猪种乳头数与产仔数的关系,发现乳头数多的母猪产仔数和断奶仔猪数也更多。因此,乳头数是生猪生产中非常重要的繁殖性状。此外,随着育种技术的应用,以丹麦大白、长白为代表的多猪种的产仔数性状得到了有效的选育提升,但其乳头数未经系统选育提升,导致仔猪数多于乳头数,会使得一部分仔猪需要通过寄养或人工哺乳的方式存活,增加生产成本和管理难度。因此,乳头数的性状改良是当下育种研究的重点之一。【前人研究进展】乳头数性状由多基因控制,遗传机理复杂。仅基于表型对乳头数性状进行选育的遗传进展较慢。结合分子标记选育能有效加快性状改良的进程[3]。SNP芯片分型技术能够高效、准确地获得大量SNPs的基因型,推动了F[4]分析和GWAS分析在鉴定复杂性状候选QTLs和基因上的应用。HE等[5]按照产仔数EBV排名选择前10%和后10%的二花脸猪进行F分析,鉴定到、和等3个潜在影响产仔数的候选基因。李开军等[6]采取相同的选择策略针对苏淮猪的中性洗涤纤维表观消化率进行F分析,鉴定到多个与纤维表观消化率性状相关的基因和SNPs。在商业猪种乳头数的GWAS分析中,MOSCATELLI等[7]使用60K芯片分型了821头意大利大白猪,在6、7号染色体上分别鉴定到显著影响前后乳头对称性以及乳头数的QTLs,并将和作为两种性状对应的候选基因。LI等[8]在杜洛克猪的GWAS分析中新发现了影响乳头数的和影响左右乳头对称性的。在国内地方猪种的GWAS分析中,TANG等[9]在白杜洛克和二花脸的杂交群体中定位到影响乳头数的和。ZHOU等[10]基于GWAS分析在1、13和X染色体上鉴定到多个与苏山猪乳头数显著关联的SNPs。目前,猪QTLdb数据库中总共收录了624个与乳头数关联的QTLs,每条染色体上均有分布(https://www. animalgenome.org/cgi-bin/QTLdb/index/)。【本研究切入点】尽管已有多数关于乳头数的研究报道,但是影响乳头数的主效基因和因果突变仍未知。已有研究主要集中在国外商业猪种,其研究结果通常存在群体异质性,而国内地方猪种或含有地方猪种血缘的培育猪种的研究有待系统开展。苏淮猪是以本研究团队作为技术支撑单位,历经12年时间,于2010年培育而成的国家级瘦肉型猪新品种,含75%的大白猪和25%的淮猪血统[11]。该群体在包括乳头数在内的性状上存在一定的变异,是鉴别影响猪乳头数QTL、候选基因以及解析乳头数变异遗传机制的良好素材。【拟解决的关键问题】本研究以苏淮猪为试验动物,统计了群体内乳头数的分布状况,并使用SNP芯片进行个体基因分型。基于SNP芯片数据估计了乳头数的遗传力,选取多乳头数个体和少乳头数个体进行F分析,并在整个群体进行了表型和基因型间的GWAS分析,结合F分析和GWAS分析挖掘显著影响乳头数的候选QTLs和基因,并对候选基因进行功能注释。本研究的结果旨在为研究猪乳头数的遗传基础提供理论参考,同时为苏淮猪乳头数的分子育种提供关键分子标记。
1 材料与方法
1.1 试验动物
试验选用335头苏淮育肥猪、374头苏淮种猪(25头公和349头母猪)共709头苏淮猪均在江苏省淮安市淮阴新淮种猪场内饲养。在2019年内,准确记录其左、右乳头数及总乳头数。苏淮育肥猪作为试验群体用于鉴定与乳头数显著关联的SNPs,全部群体用于验证部分显著的SNPs的显著性。
1.2 试验方法
1.2.1 DNA提取与基因分型 采集709头苏淮猪的耳组织并贮存于2 mL离心管中。离心管内装有2/3体积的75%酒精且外侧管壁贴有胶带以记录耳号。取小部分耳组织用于提取DNA,剩余样品保存至-20 ℃冰箱。DNA提取的试剂及步骤参见天根DNA提取试剂盒说明。试验群体中,浓度和质量符合要求的DNA进一步使用Geneseek GGP 80K芯片进行SNP分型。分型后,使用PLINK[12]软件基于以下标准进行质量控制:SNP在个体的检出率及个体的SNP检出率均大于0.9,次等位基因频率大于0.05。总共有334个个体(其中有一个个体因未达到质控标准被去掉)及50 504个SNPs通过了质控并用于后续的分析。
1.2.2 主成分分析 群体内分层可能会导致不同的亚群内部个体之间的关系比实际更紧密,进而影响总体的关联性评估。本研究使用PLINK[12]软件执行了主成分分析,计算了前10个主成分的特征值并以主成分1和主成分2分别作为横、纵坐标绘制PCA图。
1.2.3 遗传力评估 使用DMU[13]软件估计试验群体单个性状的方差组分及个体育种值。模型如下:
y=+sex+ad+e
其中,y代表左、右、总乳头数;为群体表型均值;sex为个体的性别;个体的加性效应ad,服从正态分布ad-(0,);残差项e,服从正态分布e-(0,);是由invgmatrix[14]软件使用50 504个SNPs构建的基因组亲缘关系矩阵;是单位阵;和分别为加性方差和残差方差。基因组遗传力的计算公式为:2/(+)。
1.2.4 群体分化指数分析 研究进行了试验群体内多乳头数和少乳头数两组间的F分析,以检测多乳头数个体与少乳头数的个体之间显著分化的位点。分组规则如下:首先,按照左、右、总乳头数的GEBV分别进行排序,各选择排名前10%的个体和排名后10%的个体分别作为F分析的多乳头组和少乳头组;其次,由于GEBV是模型估计的值,可能存在一定的偏差,因此剔除了多乳头组中表型值低于平均值的个体以及少乳头组中表型值高于平均值的个体。最终,左、总乳头数的多乳头组及右、总乳头数的少乳头组均有32个个体,其余组为33个个体,并使用独立样本t检验估计极端乳头组间表型和GEBV值的差异性。F分析的模型参照WEIR等[15]的研究模型并且使用VCFTOOLS[16]软件计算全部SNPs的F值并取绝对值。取F值的绝对值前5%的位点作为显著分化的位点。左、右、总乳头数F分析的阈值分别为0.1674、0.1469、0.1482,均接近WRIGHT等[4]划分的高度分化的阈值条件(0.15<F<0.25)。
1.2.5 全基因组关联分析 试验群体乳头数与全基因组50 504个SNPs间的关联性检验通过LDAK[17]软件的Wald检验实现。模型如下:
=++++
其中,为乳头数表型;为群体均值;为SNP基因型矩阵,为基因型效应值;为固定效应矩阵,为性别效应;为剩余多基因效应,服从正态分布-(0,),为校正的基因组亲缘关系矩阵;为残差,服从正态分布-(0,);其余字母含义同遗传力估计的模型。由于GWAS分析共执行了50 504次假设检验,因此,采用BONFERRONI[18]方法计算基因组显著的阈值为0.05/50504(9.90×10-7)。考虑到BONFERRONI[18]方法的严格性,染色体显著的阈值定义为1/50504(1.98×10-5)[19]。其次,在估计单个SNP效应时,GWAS模型同时添加了SNP的固定效应和多基因效应,造成了效应的重复校正。参考SCHMID等[20]的方法,即每次估计SNP效应时,剔除该SNP所在染色体,使用剩余染色体的SNPs构建矩阵。GWAS的结果包括曼哈顿图和Q-Q图由R包CMplot[21]可视化。最后,使用Haploview4.2[22]软件绘制16号染色体上6.36—10.66 Mb的候选SNPs的连锁不平衡图,且只计算1 Mb内的SNPs之间的LD值。
1.2.6 显著SNPs解释的表型方差比例 为进一步评估试验群体的GWAS分析,计算了显著SNPs所解释性状的表型方差的比例。计算公式同ZHANG等[23]:=2(1-)2/。为解释的表型方差比例;为最小等位基因频率;为SNP的效应值;为表型方差,并且代表观测值的离散程度。
1.2.7 候选SNPs所在候选基因的鉴别与功能注释 本研究使用的80K芯片上的SNPs的物理位置是根据猪11.1参考基因组注释。候选SNPs所在或者附近的基因作为候选基因。候选基因名称通过Ensembl数据库(http://www.ensembl.org/)查找。候选基因的功能使用GeneCards(https://www.genecards.org/)和Pubmed数据库(https://pubmed.ncbi.nlm.nih.gov/)注释。
1.2.8 候选SNPs与全部群体乳头数关联分析 本研究将左、右、总乳头数的F以及GWAS分析共同定位的显著SNPs作为候选SNPs。本研究选取了每条染色体上最显著的候选SNPs以验证其在全部群体中的显著性。上述SNPs的引物在NCBI的Primer-BLAST模块(https://www.ncbi.nlm.nih.gov/tools/primer-blast/)设计,引物信息如表1,PCR扩增流程见表2,扩增产物通过Sanger测序判定SNPs的基因型。

表1 候选SNPs的引物信息
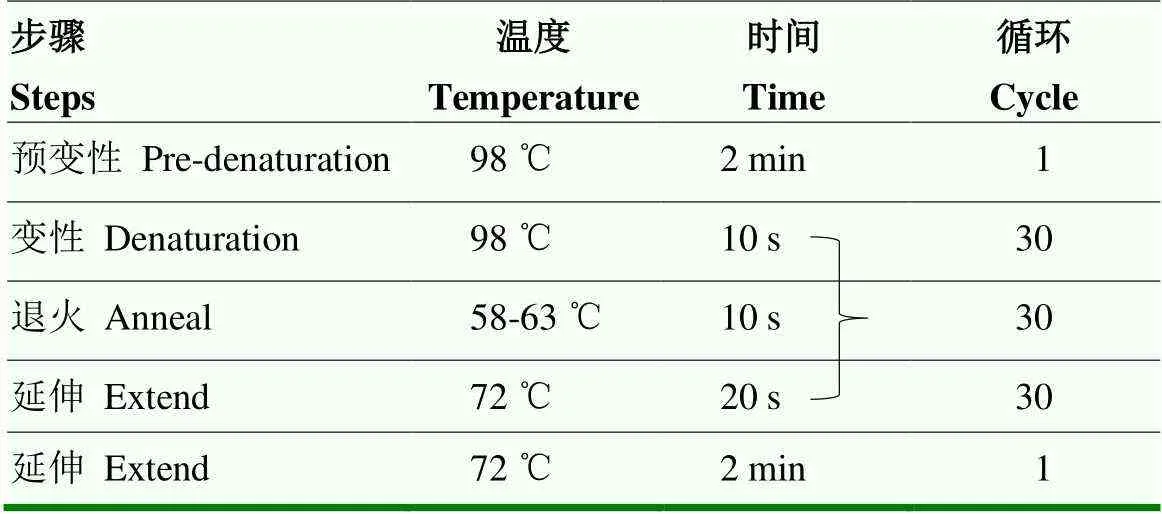
表2 PCR扩增流程
使用SAS 9.4(https://odamid-apse1.oda.sas.com/ SASStudio)的GLM模型对SNPs基因型和全部群体乳头数进行关联分析,并使用Bonferroni方法进行多重比较。模型如下:
y=+sex+g+e
其中,g为SNPs的固定效应,其余字母含义同遗传力估计的模型。
2 结果
2.1 苏淮猪试验群体乳头数描述性统计及遗传力估计
表3展示了试验群体334头苏淮猪乳头数的分布、变异情况及遗传力。左、右、总乳头数的范围分别是5—11、5—10、11—21,表型的分布比较分散。左、右、总乳头数的变异系数分别为10.20%、9.26%、8.50%。苏淮猪左、右、总乳头数的遗传力分别为0.212、0.257、0.312,属于中高等遗传力性状。
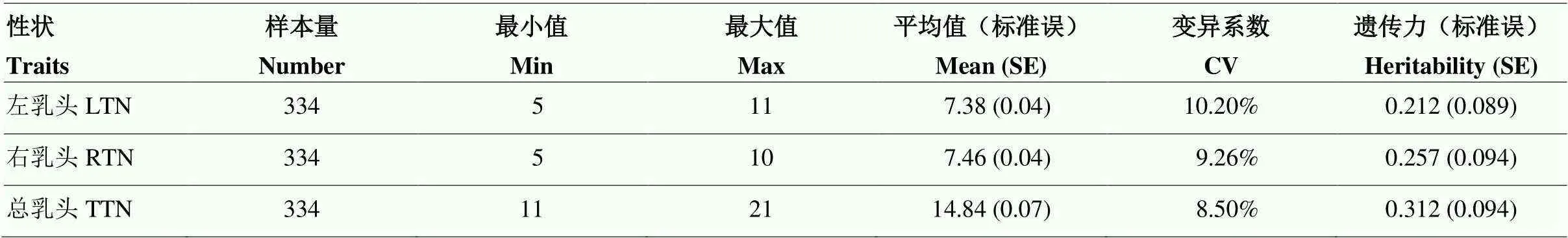
表3 334头苏淮猪乳头数描述性统计及遗传力估计
LTN,左乳头;RTN,右乳头;TTN,总乳头;SE,标准误;CV,变异系数。表7同
LTN, Left Teat Number; RTN, Right Teat Number; TTN, Total Teat Number; SE Standard Error; CV, Coefficient Variation. The same as table 7
2.2 苏淮猪试验群体分层检验
基于50 504个SNPs的基因型计算的主成分1和主成分2占总体特征值的百分比是相近的,并且群体内没有明显离群的个体(图1)。因此,334头苏淮猪的群体内不存在更小的亚群。
2.3 苏淮猪试验群体乳头数FST分析
试验群体中用于F分析的多乳头组的表型均值及GEBV均值均显著高于少乳头组(表4)。3个乳头数性状的F分析均鉴定到2 525个在多乳头组和少乳头组之间高度分化的SNPs(图2)。左乳头F值排名前五的SNPs位于SSC10的28.10—28.37 Mb;右乳头F值排名前五的SNPs位于SSC8、SSC13、SSC14上;总乳头F值排名前五的SNPs位于SSC8、SSC13、SSC16上(表5)。
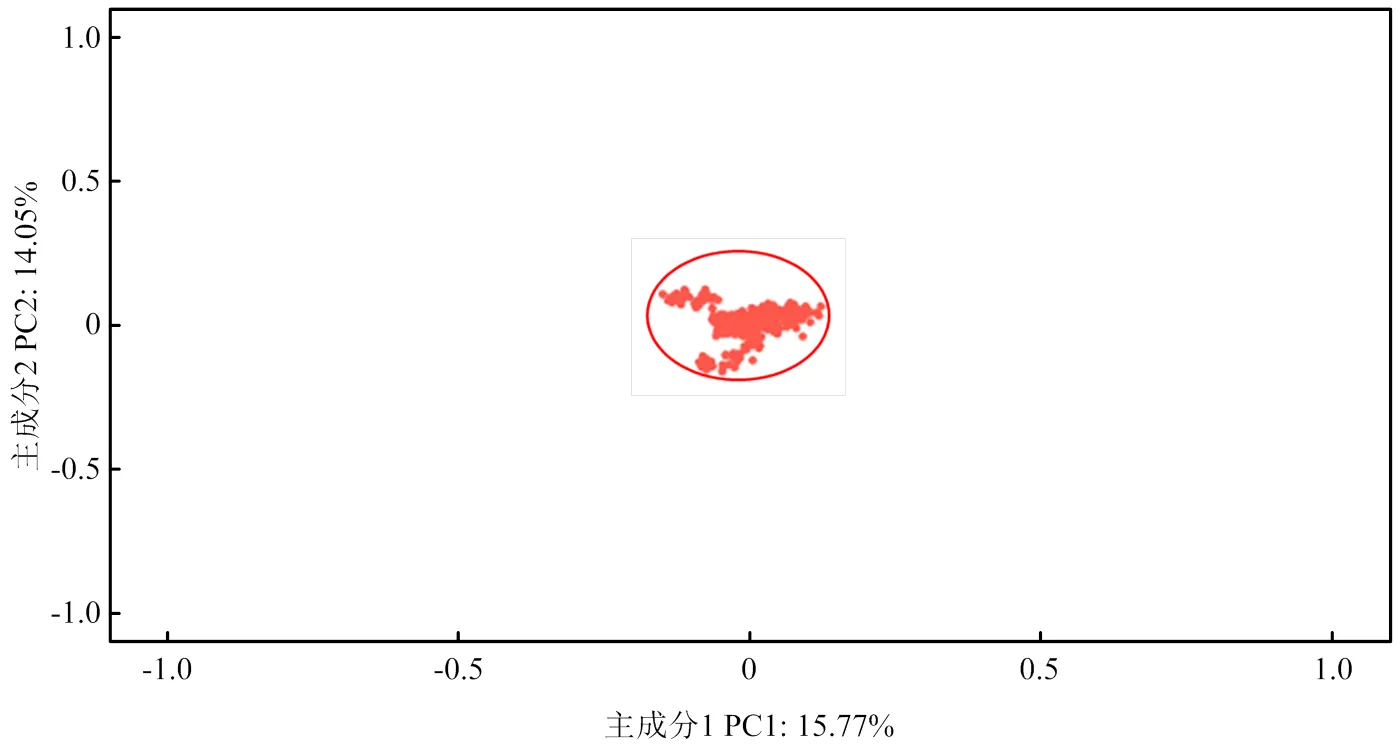
图1 334头苏淮猪的PCA图
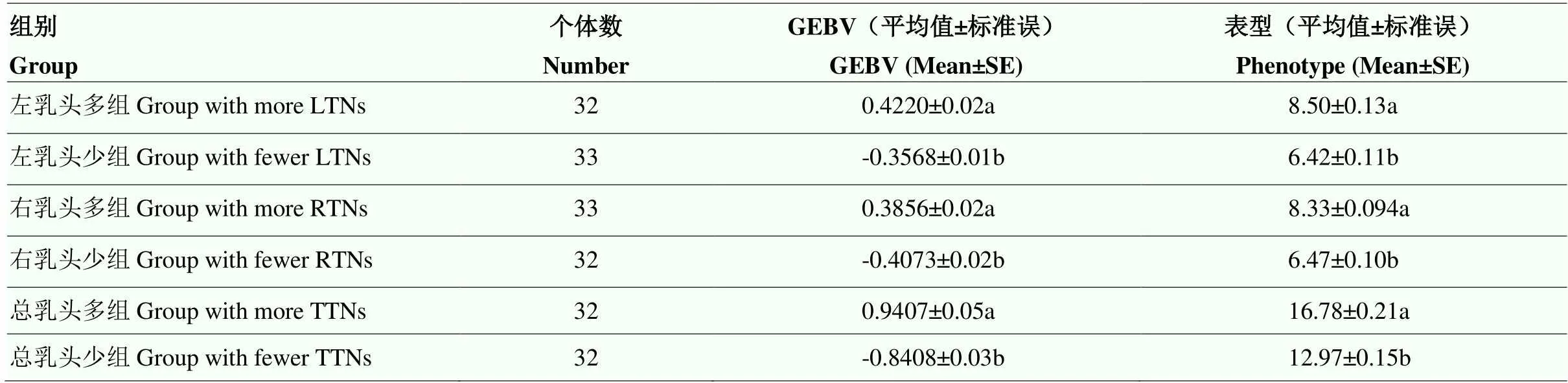
表4 极端乳头数个体GEBV及表型分布
极端乳头组间同列不同字母表示差异显著
Different letters in the same column between groups with extreme teats indicated significant differences

A:左乳头;B:右乳头;C:总乳头。X轴为1-18号染色体,Y轴为FST值,红色虚线代表阈值线

表5 苏淮猪乳头数FST值排名前五的位点
2.4 苏淮猪试验群体乳头数GWAS分析
通过GWAS分析以更精确地定位影响乳头数的位点。左、右、总乳头数的GWAS分析的基因膨胀因子分别为1.07、1.179、1.035,并且Q-Q图(图3)中的观测值和期望值没有明显的分离,说明不存在群体分层导致的膨胀现象。如表6和图3所示,GWAS分析在SSC7、SSC13、SSC16、SSC18上鉴定到了22个与乳头数显著关联的SNPs,其中16号染色体存在大量连续显著的位点。显著关联的位点中,除了rs81315330 和rs319610259,剩余SNPs的F值均超过了F分析的阈值线。
将在极端乳头数个体间高度分化且与乳头数显著关联的20个位点认定为潜在地影响乳头数的候选位点。在候选位点中,SSC13上的rs81444134以及SSC18上的rs81233299均为左乳头的候选位点,均未在之前的研究中被报道,且分别解释了6.16%和6.62%的表型方差。其中,rs81444134在右、总乳头的F分析中的F值排名前五。SSC7上的rs80894106为总乳头的候选位点,解释了8.03%的表型方差。其余的左、右、总乳头的候选位点集中位于SSC16的6.36—10.66 Mb区间内。LD分析显示,SSC16上7.47—8.27 Mb内的7个候选SNPs形成了一个795 kb的单倍型块(图4),且其中的位点rs325271937(7.99 Mb)、rs343435106(8.04 Mb)、rs336743934(8.27 Mb)均位于cadherin 18()基因上。单倍型块中的位点rs337606862(7.47 Mb,SSC16)在右、总乳头的GWAS分析中均达到了全基因组显著水平,且其右乳头以及总乳头的PVE分别为7.73%和7.57%。目前在QTL数据库中,SSC16上的7.47—8.27 Mb区间没有发现已报道的影响乳头数的QTL,因此该区域可能是一个新的影响乳头数的QTL,且rs337606862是一个重要的候选SNP。
2.5 候选基因注释
SSC16上7.47—8.27 Mb的单倍型块内包含了和cadherin 18()2个编码蛋白的基因(图4)。编码一类钙黏蛋白,与组织发育中细胞的识别、分选等有关,是潜在影响乳头数的候选基因。SSC7上的rs80894106的上游28.35 kb处为,下游5.66 kb处为,其中为已报道的影响乳头数的候选基因。距离rs81444134(SSC13)和rs81233299(SSC18)最近的编码蛋白的基因分别为golgi associated kinase 1A(,38.9 kb)和taste 2 receptor member 3(,2.5 kb),暂未发现这两个基因与乳头发育有关。
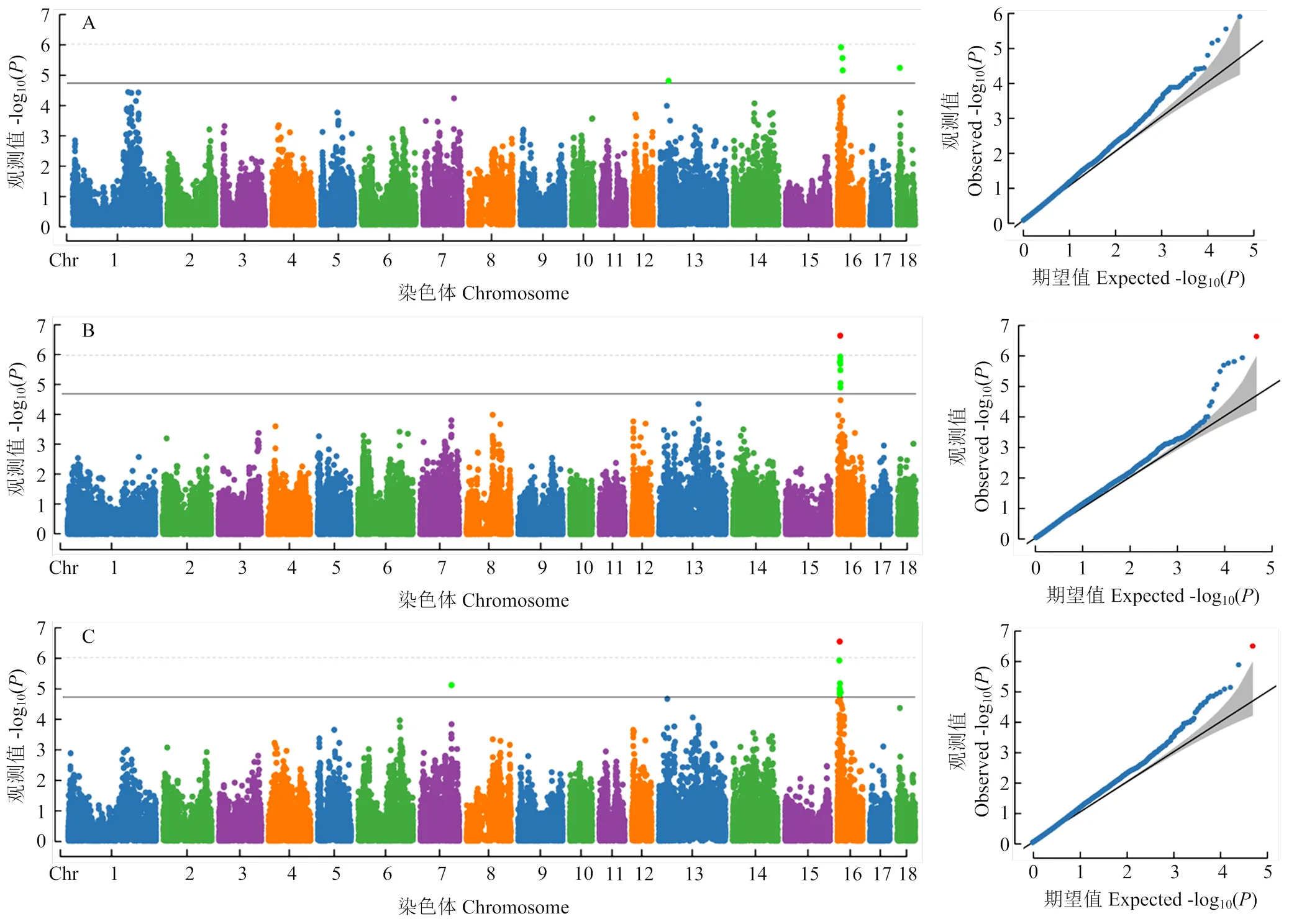
A:左乳头;B:右乳头;C:总乳头。X轴为1-18号染色体,Y轴为-log10(P)。曼哈顿图的实线和虚线分别代表染色体显著阈值线及基因组显著阈值线

方格内的数字表示两个SNP之间的D’值,深红色意味着高的连锁程度。共有两个蛋白编码基因位于795 kb的单倍型块内
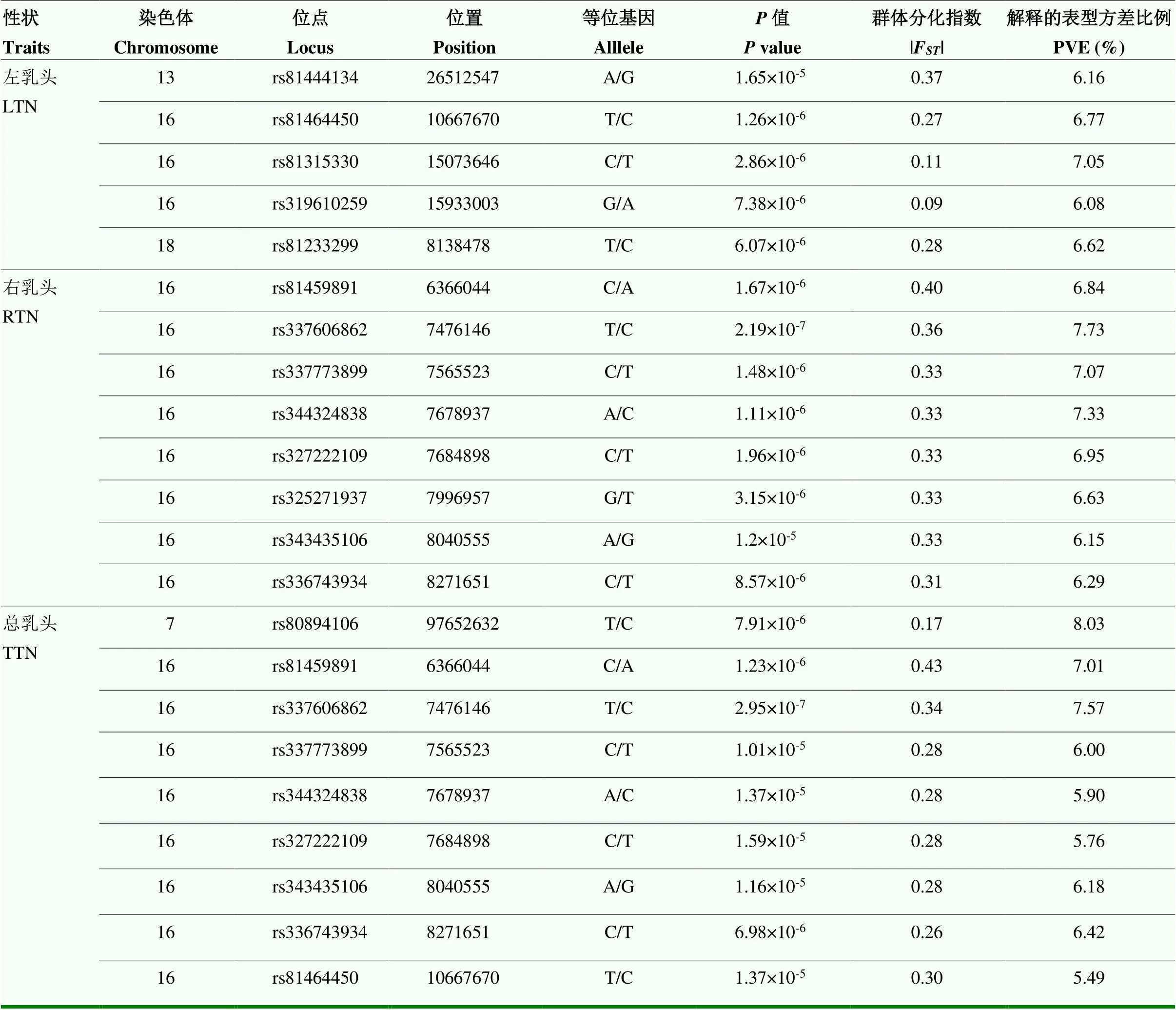
表6 苏淮猪乳头数GWAS鉴定的显著位点的详细信息
2.6 SSC7、SSC13、SSC16、SSC18上最显著位点与苏淮猪全部群体乳头数关联分析
选择4条染色体上最显著位点与709头苏淮猪的左、右、总乳头数进行关联分析。709头苏淮猪的左、右、总乳头数的变异系数分别为8.58%、8.22%和7.34%(表7)。关联分析结果如表8,4个位点均与左、右、总乳头数显著关联(<0.05),说明F和GWAS分析鉴定的候选位点在样本量更大的全部群体中仍然与乳头数具有关联性。因此,这4个位点可以作为分子标记用于苏淮猪乳头数的选育提升。
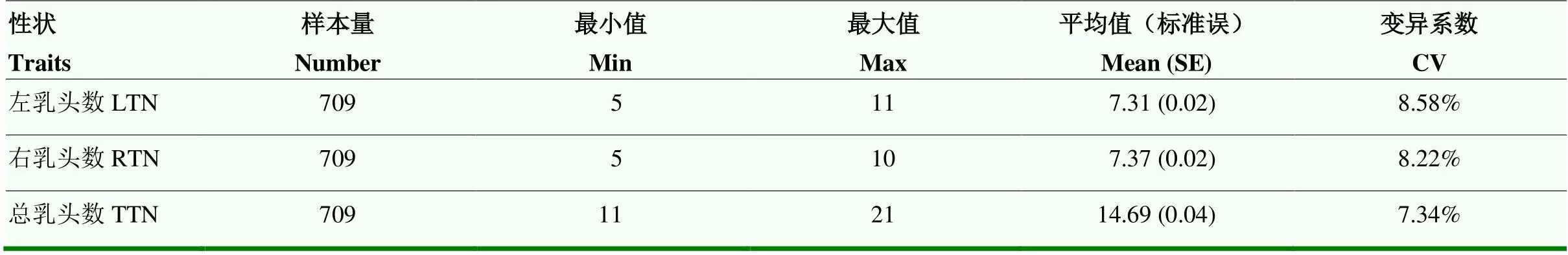
表7 709头苏淮猪乳头数描述性统计
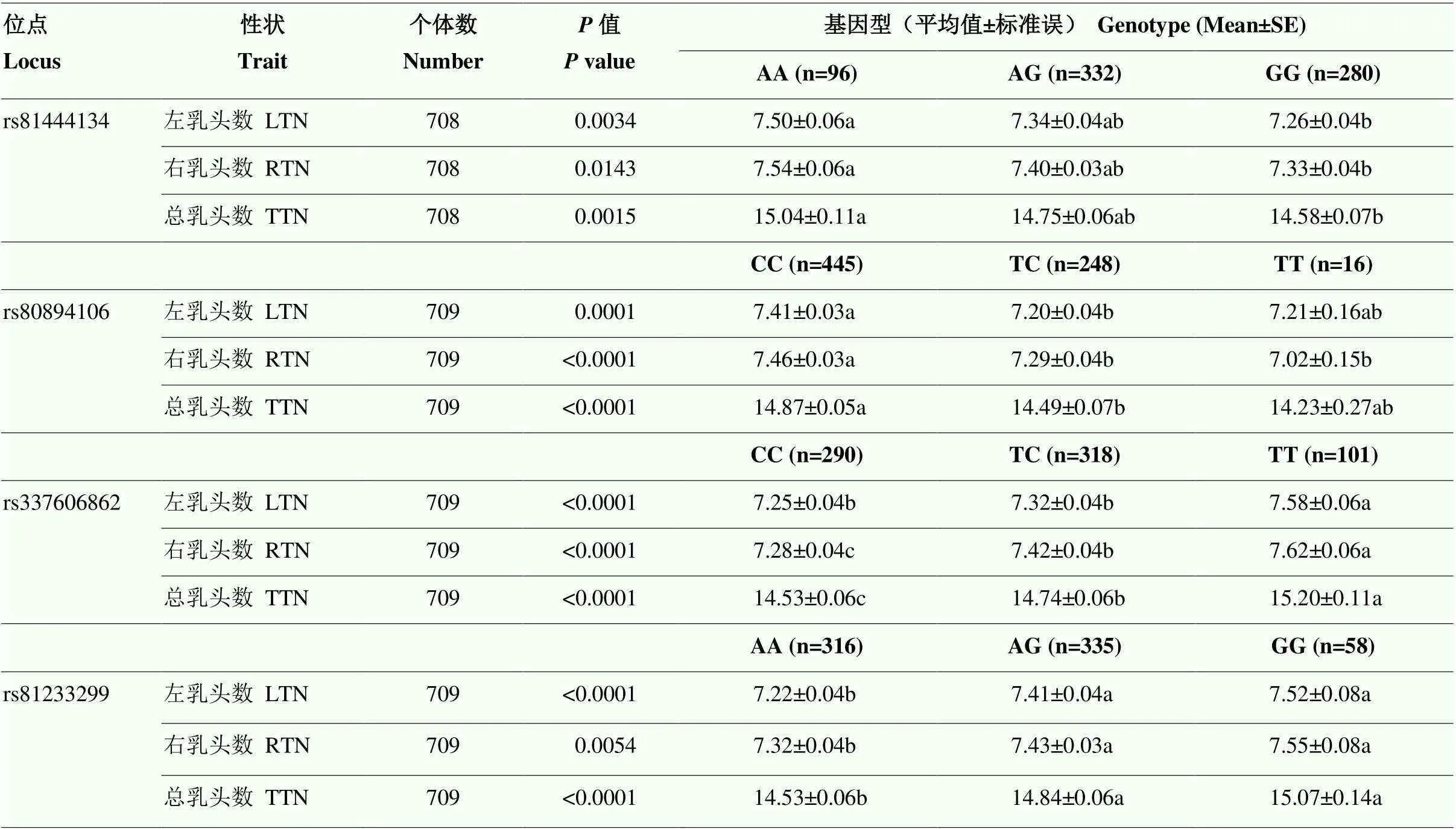
表8 4个位点与苏淮猪全部群体乳头数关联分析
同行数字上标不同字母表示差异显著(<0.05);rs81444134在80K芯片中有一个个体缺失,其有基因型的个体数为708头
Different superscripts of numbers in the same line indicate significant differences; rs81444134 has one missing individual in 80K chip, and the number of genotyped individuals is 708
3 讨论
3.1 苏淮猪乳头数变异统计及遗传力估计
苏淮猪是一个培育品种,群体内各性状可能存在一定的变异。本研究统计的334头苏淮育肥猪以及全部群体的709头苏淮猪的左、右、总乳头数的变异系数均超过了7%,这也证实了这一论述。周李生等[24]统计了中外猪种总乳头数的分布情况,丹麦、英国、美国等猪种的乳头集中在12—16个,中国猪种的乳头分布在10—22个之间。本研究的苏淮猪的总乳头在11—21个之间,说明该群体是研究乳头数变异机制的理想群体。在遗传力估计上,多数研究估计的大白、长白、杜洛克猪的总乳头数的遗传力均在0.2—0.5之间[8, 25-26],与苏淮猪遗传力的结果相近。同时,LI等[8]估计的杜洛克猪的左乳头(0.148)遗传力低于右乳头(0.177),这与苏淮猪的结果一致。
3.2 影响苏淮猪乳头数QTL的鉴定
在实际的选种中,苏淮猪育种者倾向选择多乳头的母猪留作种用,这种人工选择作用会使得有利等位基因的频率上升并在多乳头的群体和少乳头的群体之间产生基因频率的差异。因此,期望通过F分析检测极端乳头数个体之间等位基因频率差异显著的位点。由于乳头数的遗传力中等,加上群体数量有限,因此结合育种值和表型值筛选极端乳头的个体是较优的方法。同时,相较于仅依靠系谱信息的BLUP模型,基于基因组信息的GBLUP模型估计的育种值更准确[27],且广泛应用于畜禽的育种中[28]。因此,本研究结合GEBV和表型值确定极端乳头数的个体,而且统计发现本研究选择的极端乳头数组间的乳头数GEBV和表型值均存在显著差异,证明样品选择的合理性。F分析检测到的高度分化的位点在18条常染色体上均有分布。由于F分析仅选择了极端乳头数的个体,群体量较小,检测到的显著分化的位点可能含有因抽样误差产生的假阳性的结果。因此,后续又开展了试验群体的GWAS分析,并选定在F分析中高度分化且在GWAS分析中与乳头数显著关联的位点为更可能的影响乳头数的候选位点。
候选位点中,rs81444134(26.51 Mb,SSC13)和rs81233299(8.13 Mb,SSC18)均未在之前的研究中报道,是本研究新发现的乳头数的候选位点。其中,rs81444134的F值在右、总乳头的F分析中排名前五。rs81444134和rs81233299附近的基因为和,且Genecards数据库显示这两个基因分别与高尔基体激酶以及味道受体有关,但其影响乳头形成的功能有待进一步研究。在GWAS中,SSC7上97.65 Mb的候选位点rs80894106与总乳头数显著关联,且与MOSCATELLI等[7]和LI等[8]的研究中报道的影响大白猪和杜洛克猪的总乳头数的位点相一致。左、右、总乳头的GWAS均在SSC16上出现连续的信号峰,且显著信号集中在6.36—10.66 Mb。同时,3种乳头数的F和GWAS分析在该区域均有一致的信号趋势。本研究进一步对该区域进行LD分析,发现7.47—8.27 Mb区域内的候选SNPs之间高度连锁,形成了一个795 kb的单倍型块,而目前未有研究报道该单倍型区域与乳头数相关。单倍型块内的位点rs337606862与右乳头和总乳头均是最显著关联的,值分别为2.19×10-7和2.95×10-7。ZHUANG等[29]在加拿大杜洛克群体的GWAS中鉴定到SSC16上6.12 Mb处的总乳头的QTL;BIDANEL等[30]使用梅山猪和大白猪的杂交群体鉴定的乳头数的QTL位于SSC16的6.3—6.4 Mb处;这两个QTLs分别距离本研究的7.47—8.27 Mb的单倍型块约1.37 Mb和1.07 Mb远。因此,SSC16上的7.47—8.27 Mb区域应是一个新的影响乳头数的QTL,且位点rs337606862是QTL内具有较大效应的SNP。区域7.47—8.27 Mb包含了2个编码蛋白的基因,其中的1个基因暂未有文献研究,另外一个是一种钙黏蛋白基因。生成Ⅱ型的钙黏附素,在小鼠发育中的性腺细胞中高表达[31]。钙黏附素在机体发育过程,包括胚胎期参与识别与分选不同种类的细胞,在组织形成中起到重要作用[32]。而猪的乳头大概在胚胎期的23—26 d左右形成[33]。同时,钙黏附素也可以调节细胞的增殖和凋亡活动[34]并且其功能的改变可能会导致乳腺癌的发生[35]。因此,推测是一个新的影响猪乳头形成的候选基因,但该基因影响乳头形成的机制还需要进一步研究。
为了进一步验证试验群体鉴定的乳头数的候选位点的可靠性,本研究进一步选择了包含试验群体在内的共709头苏淮猪群体并进行了最显著的SNPs:rs80894106(SSC7)、rs81444134(SSC13)、rs337606862(SSC16)、rs81233299(SSC18)的基因型与苏淮猪群体乳头数的关联分析。结果表明,上述位点仍然与709头苏淮猪的乳头数显著关联。尽管试验群体使用了混合线性模型,而整个群体的关联分析由于没有足够的亲缘信息,没有考虑随机加性效应,但上述结果一定程度上验证了4个最显著位点与苏淮猪乳头数关联的可靠性,可作为乳头数选育的潜在分子标记。
4 结论
本研究发现苏淮猪群体内乳头数存在一定的变异,通过基因组F分析和GWAS分析,在苏淮猪中鉴定到20个与乳头数显著相关的位点。其中,SSC13的26.51 Mb和SSC18的8.13 Mb是新的乳头数的候选QTLs;SSC16的7.47—8.27 Mb区间存在连续与乳头数显著关联的位点,是新发现的影响乳头数的候选QTL区域,该区域内可能是新的影响猪乳头形成的候选基因。
[1] ALEXOPOULOS J G, LINES D S, HALLETT S, PLUSH K J. A review of success factors for piglet fostering in lactation. Animals: an Open Access Journal from MDPI, 2018, 8(3): 38.
[2] KIM J S, JIN D I, LEE J H, SON D S, LEE S H, YI Y J, PARK C S. Effects of teat number on litter size in gilts. Animal Reproduction Science, 2005, 90(1/2): 111-116.
[3] ROHRER G A, NONNEMAN D J. Genetic analysis of teat number in pigs reveals some developmental pathways independent of vertebra number and several loci which only affect a specific side. Genetics, Selection, Evolution: GSE, 2017, 49(1): 4.
[4] WRIGHT S. The genetical structure of populations. Annals of Eugenics, 1951, 15(4): 323-354. doi:10.1111/j.1469-1809.1949. tb02451.x.
[5] HE L C, LI P H, MA X, SUI S P, GAO S, KIM S W, GU Y Q, HUANG Y, DING N S, HUANG R H. Identification of new single nucleotide polymorphisms affecting total number born and candidate genes related to ovulation rate in Chinese Erhualian pigs. Animal Genetics, 2017, 48(1): 48-54.
[6] 李开军, 侯黎明, 蒲广, 刘航, 刘根盛, 石传宗, 金通, 周娟, 李平华, 黄瑞华. 基于全基因组Fst和nSL分析鉴别苏淮猪中性洗涤纤维表观消化率相关候选基因位点. 畜牧兽医学报, 2021, 52(7): 1809-1819.
LI K J, HOU L M, PU G, LIU H, LIU G S, SHI C Z, JIN T, ZHOU J, LI P H, HUANG R H. Identification of candidate gene loci related to apparentdigestibility of suhuai pigs based on genome-wide fst and nSLAnalyses. Acta Veterinaria et Zootechnica Sinica, 2021, 52(7): 1809-1819. (in Chinese)
[7] MOSCATELLI G, DALL'OLIO S, BOVO S, SCHIAVO G, KAZEMI H, RIBANI A, ZAMBONELLI P, TINARELLI S, GALLO M, BERTOLINI F, FONTANESI L. Genome-wide association studies for the number of teats and teat asymmetry patterns in Large White pigs. Animal Genetics, 2020, 51(4): 595-600.
[8] LI Y, PU L, SHI L Y, GAO H D, ZHANG P F, WANG L X, ZHAO F P. Revealing new candidate genes for teat number relevant traits in duroc pigs using genome-wide association studies. Animals: an Open Access Journal from MDPI, 2021, 11(3): 806.
[9] TANG J H, ZHANG Z Y, YANG B, GUO Y M, AI H S, LONG Y, SU Y, CUI L L, ZHOU L Y, WANG X P, ZHANG H, WANG C B, REN J, HUANG L S, DING N S. Identification of loci affecting teat number by genome-wide association studies on three pig populations. Asian-Australasian Journal of Animal Sciences, 2017, 30(1): 1-7.
[10] ZHOU L, ZHAO W, FU Y, FANG X, REN S, REN J. Genome-wide detection of genetic loci and candidate genes for teat number and body conformation traits at birth in Chinese Sushan pigs. Animal Genetics, 2019, 50(6): 753-756.
[11] WANG, LI, ZHOU, GAO, LIU, LI, NIU, ZHANG, LI, ZHOU, HUANG. Association of twelve candidate gene polymorphisms with the intramuscular fat content and average backfat thickness of Chinese suhuai pigs. Animals, 2019, 9(11): 858.
[12] PURCELL S, NEALE B, TODD-BROWN K, THOMAS L, FERREIRA M A R, BENDER D, MALLER J, SKLAR P, DE BAKKER P I W, DALY M J, SHAM P C. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics, 2007, 81(3): 559-575.
[13] MADSEN P, JENSEN J. A user’s guide to DMU. A package for analysing multivariate mixed models version, 2013, 6: 1-33.
[14] VANRADEN P M. Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008, 91(11): 4414-4423.
[15] WEIR B S, COCKERHAM C C. Estimating f-statistics for the analysis of population structure. Evolution; International Journal of Organic Evolution, 1984, 38(6): 1358-1370.
[16] DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T, MCVEAN G, DURBIN R, GENOMES PROJECT ANALYSIS GROUP 1 0 0 0. The variant call format and VCFtools. Bioinformatics (Oxford, England), 2011, 27(15): 2156-2158.
[17] SPEED D, HEMANI G, JOHNSON M R, BALDING D J. Improved heritability estimation from genome-wide SNPs. The American Journal of Human Genetics, 2012, 91(6): 1011-1021.
[18] YANG Q, CUI J, CHAZARO I, CUPPLES L A, DEMISSIE S. Power and type I error rate of false discovery rate approaches in genome- wide association studies. BMC Genetics, 2005, 6(Suppl. 1): S134.
[19] YE S P, CHEN Z T, ZHENG R R, DIAO S Q, TENG J Y, YUAN X L, ZHANG H, CHEN Z M, ZHANG X Q, LI J Q, ZHANG Z. New insights from imputed whole-genome sequence-based genome-wide association analysis and transcriptome analysis: the genetic mechanisms underlying residual feed intake in chickens. Frontiers in Genetics, 2020, 11: 243.
[20] SCHMID M, MAUSHAMMER M, PREUß S, BENNEWITZ J. Mapping QTL for production traits in segregating Piétrain pig populations using genome-wide association study results of F2crosses. Animal Genetics, 2018, 49(4): 317-320.
[21] YIN L L, ZHANG H H, TANG Z S, XU J Y, YIN D, ZHANG Z W, YUAN X H, ZHU M J, ZHAO S H, LI X Y, LIU X L. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics, Proteomics & Bioinformatics, 2021, 19(4): 619-628.
[22] BARRETT J C, FRY B, MALLER J, DALY M J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 2004, 21(2): 263-265.
[23] ZHANG T Y, GAO H D, SAHANA G, ZAN Y J, FAN H Y, LIU J X, SHI L Y, WANG H W, DU L X, WANG L X, ZHAO F P. Genome-wide association studies revealed candidate genes for tail fat deposition and body size in the Hulun Buir sheep. Journal of Animal Breeding and Genetics = Zeitschrift Fur Tierzuchtung Und Zuchtungsbiologie, 2019, 136(5): 362-370.
[24] 周李生, 赵为民, 涂枫, 吴云鹤, 任守文, 方晓敏. 猪乳头性状生理学和遗传学研究进展. 遗传, 2019, 41(5): 384-390.
ZHOU L S, ZHAO W M, TU F, WU Y H, REN S W, FANG X M. Physiology and genetics research progress of teat traits in pigs. Hereditas, 2019, 41(5): 384-390. (in Chinese)
[25] SON M V, LOPES M S, MARTELL H J, DERKS M F L, GANGSEI L E, KONGSRO J, WASS M N, GRINDFLEK E H, HARLIZIUS B. A QTL for number of teats shows breed specific effects on number of vertebrae in pigs: bridging the gap between molecular and quantitative genetics. Frontiers in Genetics, 2019, 10: 272.
[26] TAN C, WU Z F, REN J L, HUANG Z L, LIU D W, HE X Y, PRAKAPENKA D, ZHANG R, LI N, DA Y, HU X X. Genome-wide association study and accuracy of genomic prediction for teat number in Duroc pigs using genotyping-by-sequencing. Genetics, Selection, Evolution: GSE, 2017, 49(1): 35.
[27] 李晶, 王杰, 康慧敏, 刘冉冉, 李华, 赵桂苹. 基于BLUP和GBLUP方法估计北京油鸡胴体和肉质性状遗传参数的差异. 畜牧兽医学报, 2020, 51(1): 35-42.
LI J, WANG J, KANG H M, LIU R R, LI H, ZHAO G P. The difference of genetic parameters for carcass and meat quality traits by BLUP and GBLUP methods in Beijing You chicken. Acta Veterinaria et Zootechnica Sinica, 2020, 51(1): 35-42. (in Chinese)
[28] 朱墨, 郑麦青, 崔焕先, 赵桂苹, 刘杨. 基于GBLUP和BayesB方法对肉鸡屠宰性状基因组预测准确性的比较. 中国农业科学, 2021, 54(23): 5125-5131. doi: 10.3864/j.issn.0578-1752.2021.23.016.
ZHU M, ZHENG M Q, CUI H X, ZHAO G P, LIU Y. Comparison of genomic prediction accuracy for meat type chicken carcass traits based on GBLUP and BayesB method. Scientia Agricultura Sinica, 2021, 54(23): 5125-5131. doi: 10.3864/j.issn.0578-1752.2021.23.016. (in Chinese)
[29] ZHUANG Z W, DING R R, PENG L L, WU J, YE Y, ZHOU S P, WANG X W, QUAN J P, ZHENG E Q, CAI G Y, HUANG W, YANG J, WU Z F. Genome-wide association analyses identify known and novel loci for teat number in Duroc pigs using single-locus and multi-locus models. BMC Genomics, 2020, 21: 344.
[30] BIDANEL J P, ROSENDO A, IANNUCCELLI N, RIQUET J, GILBERT H, CARITEZ J C, BILLON Y, AMIGUES Y, PRUNIER A, MILAN D. Detection of quantitative trait loci for teat number and female reproductive traits in Meishan × Large White F2pigs. Animal, 2008, 2(6): 813-820.
[31] PIPREK R P, KOLASA M, PODKOWA D, KLOC M, KUBIAK J Z. Cell adhesion molecules expression pattern indicates that somatic cells arbitrate gonadal sex of differentiating bipotential fetal mouse gonad. Mechanisms of Development, 2017, 147: 17-27.
[32] HALBLEIB J M, NELSON W J. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes & Development, 2006, 20(23): 3199-3214.
[33] ROBINSON G W. Cooperation of signalling pathways in embryonic mammary gland development. Nature Reviews Genetics, 2007, 8(12): 963-972.
[34] PIPREK R P, KLOC M, MIZIA P, KUBIAK J Z. The central role of cadherins in gonad development, reproduction, and fertility. International Journal of Molecular Sciences, 2020, 21(21): 8264.
[35] ASHAIE M A, CHOWDHURY E H. Cadherins: the superfamily critically involved in breast cancer. Current Pharmaceutical Design, 2016, 22(5): 616-638.
Identifying Quantitative Trait Loci Associated with Teat Number of Pig by Genomic Analysis

1Institute of Swine Science, Nanjing Agricultural University, Nanjing 210095;2Huaian Academy, Nanjing Agricultural University, Huai’an 223001, Jiangsu;3Huaiyin Xinhuai Pig Breeding Farm of Huai’an City, Huai’an 223322, Jiangsu
【Objective】The purposes of this study were to analyze the variation of teat number, to explore the quantitative trait locus (QTL) and candidate genes related to teat number, and to provide important molecular markers for the breeding of pig teat number.【Method】This study accurately measured left, right, total teat number of 709 Suhuai pigs (335 fattening pigs and 374 breeding pigs). Fattening pigs were selected for 80K chip genotyping and the heritability and genomic estimated breeding value (GEBV) of left, right and total teat number were calculated by chip data. Based on the rank of GEBV and phenotype of teat number, the top 10% individuals and the bottom 10% individuals were selected for Fixation Index (F) analysis to detect highly differentiated loci. Then, the loci associated with teat number were identified by genome wide association analysis (GWAS) and loci which were highly differentiated and significantly associated with teat number were selected as candidate loci.Genes located near candidate loci and related to teat number after functional annotation were selected as candidate genes.Finally,the association analyses between the most significant candidate loci on each chromosome and teat number of 709 Suhuai pigs were performed to verify the significance of the above loci.【Result】The variation coefficients of left, right and total teat number of Suhuai fattening pigs were 10.20%, 9.26% and 8.50%, respectively, and the heritability were 0.212, 0.257 and 0.312, respectively. Based onFand GWAS analyses, a total of 20 candidate loci onchromosomes (SSC) 7, 13, 16 and 18 for teat number were identified and these candidate loci could explain 5.49%-8.03% of the phenotypic variance. Among them, locus rs80894106 on SSC7 associated with total teat number was consistent with the reported candidate locus of total teat number based on Large white and Duroc pig populations, but candidate loci rs81444134 (26.51 Mb, SSC13) and rs81233299 (8.13 Mb, SSC18) of left teat number were newly discovered loci related to teat number. Interestingly, candidate loci of left, right and total teat number were mainly concentrated in the 6.36-10.66 Mb interval on SSC16;Linkage disequilibrium (LD) analysis found that candidateloci in 7.47-8.27 Mb interval fit into a 795 kb haplotype block, andthis haplotype block was a newly discovered candidate area that affected teat number;rs337606862 (7.47 Mb) in the haplotype block was the most significantly SNP associated with the left and total teat number, and three loci in the haplotype block were all located on the intron of cadherin 18 () gene;gene encoded type II cadherin, and cadherin was related to the identification, sorting, proliferation, apoptosis of cells in developing tissue and the occurrence of breast cancer. Thus,might be a new candidate gene that affected pig teat number.In addition, the most significant loci rs81444134, rs80894106, rs337606862 and rs81233299 on 4 chromosomes were genotyped in 709 Suhuai pigs in this study. After association analysis, these loci were significantly associated with teat number, and could be used as potential molecular markers for the selection of teat number.【Conclusion】In this study, 20 loci significantly related to teat number were identified in Suhuai pig population by genome analysis. Among them, 26.51 Mb on SSC13 and 8.13 Mb on SSC18 were new candidate QTLs for teat number. The 7.47-8.27 Mb on SSC16 was also a newly discovered candidate QTL for teat number, andgene in this interval might be a new candidate gene that affected the formation of pig teat.
F; GWAS; QTL; Suhuai pig; teat number; candidate genes

10.3864/j.issn.0578-1752.2023.10.014
2021-12-17;
2022-01-19
国家自然科学基金-河南联合基金项目(U1904115)、江苏现代农业(生猪)产业技术体系建设项目(JATS[2020]399)、农业农村部农业品种改良提升专项(19210387)、江苏省农业新品种创制重点项目(PZCZ201732)
尹彦镇,Tel:13851711578;E-mail:2019105013@njau.edu.cn。通信作者李平华,E-mail:lipinghua718@njau.edu.cn。通信作者黄瑞华,E-mail:rhhuang@njau.edu.cn
(责任编辑 林鉴非)
猜你喜欢
养猪(2022年4期)2022-08-17
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
现代园艺(2017年21期)2018-01-03
解放军健康(2017年5期)2017-08-01
妈妈宝宝(2017年3期)2017-02-21
湖北畜牧兽医(2015年11期)2016-01-11
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
山东农业科学(2014年1期)2015-03-09
