
基于炔丙基与丁二炔生成第一个碳环的反应机理研究
2023-08-01 06:13毛仕迪杨喜历陈朝辉孟丽苹
燃料化学学报 2023年4期
张 韦,宁 硕,毛仕迪,杨喜历,陈朝辉,孟丽苹
(昆明理工大学 云南省内燃机重点实验室,云南 昆明 650500)
多环芳烃(PAHs)是典型的环境污染物,来自有机材料(煤、石油、天然气、木材等)的不完全燃烧,是碳烟(soot)的前驱物[1,2]。PAHs 具有强致癌性和高毒性,严重地威胁着人类健康和大气环境[3]。然而,当前研究对于PAHs 的生长及soot 的形成所涉及的化学反应机理的认识仍不够完善。从分子水平了解PAHs 和soot 生成机理的化学过程,不但有利于预测燃烧过程中污染物的排放量及种类,而且对制定污染物的控制策略至关重要。碳氢燃料燃烧形成第一个碳环分子,是PAHs 生长和soot 形成的重要速率控制步[4]。因此,探究第一碳环的形成过程,对抑制发动机缸内燃烧过程中PAHs 和soot 的生成具有重要意义。
通过实验观测到微观化学计算,研究人员认为,苯或苯基是燃料燃烧过程中形成的第一个碳环[5-7]。Cole 等[8]认为,苯环的形成主要由C2Hx+C4Hx的加成反应构成。但Miller 等[9]指出单靠C2Hx+C4Hx反应生成苯的速率太慢,难以解释火焰中苯的浓度。Georgievskii 等[10]通过进一步研究发现,炔丙基(C3H3)自由基的重组是苯或苯基形成的主要途径,且燃料组分差异导致存在其他反应途径。因此,碳氢燃料燃烧过程中形成的第一个碳环分子不仅是苯或苯基,还包括其他碳环(五、六、七碳环)分子可作为PAH 和soot 的前驱物。
Hansen 等[11]提出在五元环(环戊二烯)上添加乙炔基,绕过苯基后扩环异构化形成苄基。Liu等[12]研究环戊二烯基的重组过程,发现在该过程中需要通过H、OH 等分子激活为稳定的自由基,与其他中间体结合并芳构化为稳定的萘。Trogolo等[13]通过CBS-QB3 方法计算1,3-丁二烯与C3H3的加成反应势能面,能够形成五、六和七元环的产物。Jin 等[14]通过实验和密度泛函(DFT) 模拟计算,提出丁二炔(C4H2) 连续加成和环化反应机制(CBAC),丰富了PAHs 的生长途径。然而,鉴于火焰中前驱物的多样性和化学反应的复杂性,第一碳环的生长机理还有待完善和补充。目前有研究表明,在预混合乙炔、苯、甲苯或汽油火焰的氧化区中C3H3和一些聚炔具有相当高的浓度[15-18]。C4H2是浓度最高的聚炔分子,并且C4H2分子结构中同时含有两个三键,是形成PAHs 的关键分子;而C3H3是一种共振稳定自由基,可加成生成苯,在碳氢化合物生长过程中发挥重要作用。综上所述,C3H3+C4H2可能是第一碳环形成的潜在途径,因为在加成反应过程中不需借助环境中的其他自由基脱H 形成稳定的物种,且在火焰环境中含量丰富。因此,有必要开展C3H3+C4H2的反应研究,对第一碳环的生长路径和化学动力学模型进行补充和完善。
为了完善第一个碳环分子的生长过程,本研究计算了C3H3与C4H2的加成反应路径和动力学参数。首先采用平均局部离子化能(ALIE)和静电势(ESP)结合的方法,预测C3H3与C4H2较容易反应的位点,然后基于DFT 开展反应路径计算,详细分析不同反应路径之间的竞争关系。最后,利用过渡态(TST)方法求取相关化学反应的动力学参数和反应速率常数。在分子层面深入地揭示C3H3与C4H2生成第一个碳环的微观机理变换,为构建更精确的PAHs 生长模型提供理论依据。
1 计算方法
1.1 反应位点预测
初始化学反应阶段,分子范德华表面上的ESP 分布被用来预测亲电与亲核反应位点[19]。ESP描述了位于某一点r的单位正电荷与当前体系的相互作用能。ESP 由原子核电荷Vnuc(r)和电子密度Vele(r) 组成,前者贡献正值,后者贡献负值[20]。
式中,Z是原子A 的核电荷,R代表原子核坐标,ρ是电子密度。ESP 越正(越负)的区域越有可能吸引亲核(亲电)物质的进攻,从而发生反应。距离分子表面ESP 最小点位置的最近原子,是最可能与自由基反应的位置。然而仅基于分子表面ESP预测反应位点并不可靠。将ESP 与ALIE 相结合分析预测分子反应位点会更为准确。ALIE 是从系统空间R中移除电子所需的能量,其表达式为[21]:
式中,ρi(r)是R处第i个分子轨道的电子密度;|εi|是第i个分子轨道能量的绝对值,为i轨道电子的离子化能;ρ(r) 是r处总电子密度。(r)数值 越低,表明此处的电子被束缚得越弱,因此,活性越高,越容易参与亲电或自由基反应。本研究中所有的反应活性位点预测,均使用波函数分析程序Multiwfn 结合可视化程序VMD 分析和绘制。
1.2 密度泛函理论计算
本研究所有电子结构的计算都是通过Gaussian 16[22]程序包完成,这也是量子化学计算最常用的软件[23,24]。本研究选用M06-2X[25]方法结合6-311 +G(d,p)基组进行反应物、过渡态、产物的几何结构优化和频率计算,频率校正因子为0.983[26]。通过频率分析,所有的极小点不存在虚频,而过渡态有且仅有一个虚频。采用内禀反应坐标(IRC)计算,可以验证过渡态正确连接反应物和产物。为保证能垒的准确性,本研究采用M06-2X/def2-TZVPP方法进行单点能计算。势能面(PES)上的相对能量为零点能与单点能之和。
1.3 反应速率常数计算
基于TST 理论,从Gaussian 输出的文件中获取所需参数(分子能量、自旋多重度、惯性矩等),使用Kisthelp[27]软件计算所有反应的速率常数。KisThelp 的高压极限速率常数计算公式:
式中,Cw是隧道校正因子,σ是反应路径简并度,V≠是 包含零点能的活化能垒,ΦR(T)是总配分函数,QTS(T)是过渡态配分函数。由于反应物处于热力学平衡状态,隧道效应通常会增加K值,因此,使用Wigner 方法用于获取隧道修正系数。Wigner校正因子的表达式为[28]:
式中,v†为 过渡态虚频的量级,KB玻尔兹曼常数,h普朗克常数,T为温度。
计算反应速率常数的温度区间设置为300-2500 K。速率常数的温度依赖性可以用三参数常规阿伦尼乌斯方程来表示[29]:
式中,A为指前因子;n为温度指数;Ea为活化能。
2 结果与讨论
2.1 基于ESP 与ALIE 的反应位点预测
利用ESP 与ALIE 预测反应物的活性位点,确定较容易发生反应的活性位。图1 为C3H3、C4H2的反应位点预测图。图1(a)显示,蓝色区域为较低的ALIE 值,在此区域内反应更容易发生。整个分子的较低ALIE 值(8.38、8.74 eV)是亲电位置,这表明,C3H3首尾两端的C 原子与中间C 原子相比处于弱束缚状态。因此,首尾两端的C 原子端具有更高的活性,并且容易参与亲电或自由基反应。图1(b) 为C3H3分子的ESP 表面图,其中,红色区域表示较高的ESP 值,这意味着该区域难以发生反应。蓝色区域表示ESP 为负值,表明电子电荷占主导地位。C3H3分子表面的ESP 负值区域位于首尾两端的C 原子,说明这两个位置更易发生反应。整个C3H3分子的最低ESP 值(-0.02 eV)是亲电位置,这表明,首尾两端的C 原子与中间C原子相比属于弱束缚状态。因此,首尾两端的C 原子具有更高的活性,更容易参与亲电或自由基反应。综合C3H3的ESP 和ALIE 分析,C1 和C2原子具有更强的反应活性,是与其他物质发生反应的活性位。

图1 C3H3 与C4H2 的反应位点预测图Figure 1 Prediction of reaction sites of C3H3 and C4H2 (cyan is C atom,white is H atom)
图1(c) 为C4H2分子的ALIE 图,蓝色区域为反应较为容易发生的区域,红色区域为反应难以发生的区域。两端的C 原子(10.78、10.78 eV)位置为反应相对容易发生的位点,再由图1(d)C4H2的ESP 表面可知,中间的两个C 原子位点的活性较弱,两端C 原子较容易发生亲电或亲自由基反应,电势均为-0.48 eV,是整个ESP 表面的最小值点。综合C4H2的ALIE 和ESP 分析,C4H2两端的C 原子相比中间的C 原子是处于弱束缚状态的,具有更高的反应活性,更易与其他物质发生反应。因此,本研究选取C3H3首尾两端的C 原子与C4H2发生反应,由于C4H2分子具有对称性,两端的C 原子活性最强,这两个C 原子与C3H3反应历程是一致的。因此,本研究的反应路径分为两条,一条是C3H3的C1 原子与C4H2端点的C 原子反应;另一条为C3H3的C2 原子与C4H2端点的C原子反应。
2.2 基于DFT 的反应路径分析
基于ALIE 和ESP 的反应位点预测结果,对C3H3的C1 和C2 原子与C4H2的加成反应进行计算,所有的中间体用CS,过渡态用TS 统一编号。
C3H3(C1)+C4H2的反应路径的中间体、过渡态、产物几何结构如图2(a) 所示,反应PES 如图2(b)所示。
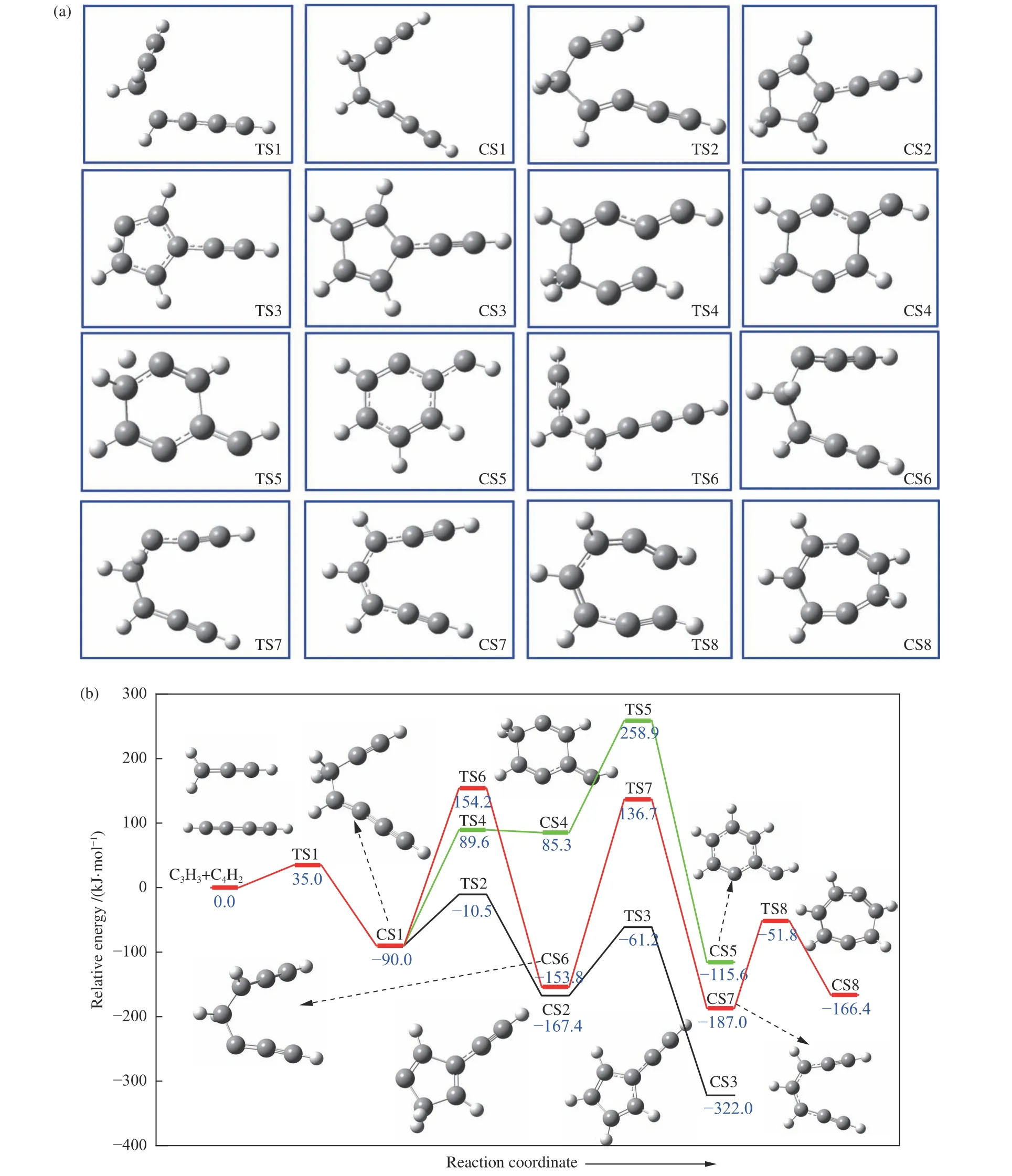
图2 (a)C3H3(C1)+C4H2 反应路径中反应物、产物、中间体和过渡态的几何构型,(b)C3H3(C1)+C4H2 势能面Figure 2 (a) Geometry of reactants,products,intermediates and transition states in C3H3(C1)+C4H2 reaction path;(b) PES of C3H3(C1)+C4H2 (Black line is path 1,green line is path 2,and red line is path 3)
首先,C3H3上 的C1 原子与C4H2分子发生加成反应,形成化合物CS1,反应能垒为35.0 kJ/mol,放出热量90 kJ/mol。C4H2(C1)+C3H3→CS1 的计算值与文献[14] 的模拟结果相近,在文献[14]中C4H2(C1)+C3H3的入口反应能垒为37.2 kJ/mol,释放94.5 kJ/mol 的热量。其中,入口反应能垒值仅相差2.2 kJ/mol,释放热量相差4.5 kJ/mol,这是由于计算基组不同所产生的差异。随后CS1 分为三条反应路径,分别生成五、六、七元环三种不同的碳环分子。在反应路径1 中,CS1 首先克服79.5 kJ/mol的能垒,发生闭环反应,形成稳定的五元环结构CS2。下一步反应为五元环C 原子上的H 迁移到邻位不饱和的C 原子上,形成富文烯基CS3,该反应需要克服106.2 kJ/mol 的能垒,释放出154.6 kJ/mol的热量。化合物CS3 为双共振稳定自由基结构,具有较强的稳定性和抗氧化性,被认为是soot 的重要前驱物[30]。
此外,CS1 可通过反应路径2 形成六元环结构CS5。首先CS1 直接发生闭环反应,形成六元环结构CS4,该反应需要克服179.6 kJ/mol 的能垒,同时吸收175.3 kJ/mol 热量。该过程为吸热反应,容易发生逆反应。随后,CS4 进行H 迁移反应,通过六元环饱和C 原子上的H 迁移到邻位不饱和C 原子上,形成稳定的六元环化合物CS5,该过程需要克服173.6 kJ/mol 能垒,并释放200.9 kJ/mol热量。
CS1 可通过反应路径3,先发生H 迁移反应,将C3H3上的H 原子迁移到邻位C4H2分子的C 原子位上,需要克服244.2 kJ/mol 的能垒,并释放63.8 kJ/mol 的热量,形成更为稳定的物质CS6。随后,再一次发生H 迁移反应,将脂肪链C 原子上的H原子迁移到邻位不饱和C 原子上,克服290.5 kJ/mol的能垒,形成化合物CS7。该反应需要较高的活化能,在化学动力学上不受支持,反应较难发生。随后直接发生闭环反应,形成七元环CS8。根据多步基元反应的限速步[31]定义可知,在C3H3(C1) +C4H2反应反应体系中,路径1-路径3 的限速步分别为CS1→TS2(79.5 kJ/mol)、CS1→TS5(348.9 kJ/mol)和CS1→TS6(244.2 kJ/mol)。由限速反应可知,路径1 在该反应体中具有较强的竞争优势,而路径2 的反应能垒高,反应较难发生,且与路径1、3 差异较大,因此,路径2 在该反应体系中竞争性较弱。就反应能垒而言,其竞争关系为:路径1 > 路径3 > 路径2。
C3H3(C2)+C4H2的反应路径的中间体、过渡态、产物几何结构如图3(a) 所示,反应PES 如图3(b) 所示。首先,C3H3上的C2 原子与C4H2分子发生加成反应,形成化合物CS9,该反应的能垒为44.1 kJ/mol,释放热量118.7 kJ/mol。随后,CS9 的C-C 键旋转,形成中间体CS10。在之后的反应路径中CS10 可通过3 条路径生成五、六、七元环。
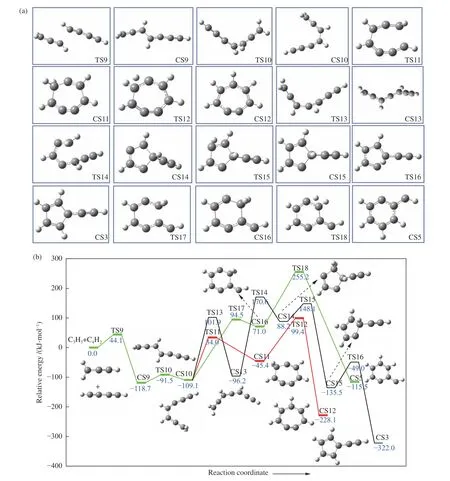
图3 (a)C3H3(C2)+C4H2 反应路径中反应物、产物、中间体和过渡态的几何构型;(b)C3H3(C2)+C4H2 势能面Figure 3 (a) Geometry of reactants,products,intermediates and transition states in C3H3(C2)+C4H2 reaction path;(b) PES of C3H3(C2)+C4H2 (Green line is path 4,red line is path 5,and black line is path 6)
在反应路径4 中,CS10 直接进行闭环反应形成七元环,该反应需要克服143.1 kJ/mol 的能垒,吸收63.7 kJ/mol 热量。在克服144.8 kJ/mol 的能垒之后,七元环上的H 原子发生迁移反应,生成稳定的中间体CS12。在反应路径5 中,CS10 的C-C支链发生旋转,需克服211 kJ/mol 的能垒,形成中间体CS13,为后续的闭环反应提供便利。接下来的反应为闭环反应,形成五元环结构的化合物CS14。CS13→CS14 克服266.8 kJ/mol 的高能垒,反应发生较为困难。随后,CS15 的五元环C 原子上的H 迁移到邻位的C 原子上形成CS15,之后再一次发生H 迁移反应,生成富文烯基CS3。在反应路径6 中,CS10 主要经历了一次环化反应和H 原子迁移反应生成六元环CS5。其中,闭环反应的反应能垒偏高,为203.6kJ/mol,随后通过发生H 迁移反应,生成稳定的六元环CS5。在C3H3(C2)+C4H2反应体系中,路径4-路径6 的限速反应为CS9→TS12(218.1 kJ/mol)、CS9→TS14(289.3 kJ/mol)、CS9→TS18(373.9 kJ/mol)。因此,通过对比反应能垒可知,其竞争关系为,路径4>路径5>路径6。
分析上述六条反应路径可知,C3H3(C2)+C4H2的反应能形成五、六、七元环的产物。其中,主要涉及到的反应有加成反应、H 迁移反应和闭环反应三种。其中,加成反应为,C3H3(C1)+C4H2→CS1(35.0 kJ/mol)与C3H3(C2)+C4H2→CS9(44.1 kJ/mol)。导致加成反应能垒有所差异的原因为前者为加成的反应位置为C3H3的C-C 单键,而后者加成反应位置为C3H3的C-C 三键。由于C-C 三键比C-C 双键更具稳定性,因此,其加成反应能垒比C-C 单键高。
就H 迁移反应而言,在整个反应体系中,存在多次H 迁移反应,可归类为以下四种:一是五元环C原子上H迁移到五元环(CS2→CS3,106.2 kJ/mol、CS14→CS15,59.9 kJ/mol、CS15→CS3,86.5 kJ/mol);二是六元环C 原子上H 原子迁移到六元环(CS4→CS5、CS16→CS5,173.6 kJ/mol、184.2 kJ/mol);三是七元环C 原子上H 原子迁移到七元环(CS11→CS12,144.8 kJ/mol);四是脂肪链上H 原子迁移到脂肪链上(CS10→CS13、CS1→CS6、CS6→CS7,211 kJ/mol、244.2 kJ/mol、290.5 kJ/mol)。通过对比可知,碳环分子上的H 迁移反应能垒取决于碳环上C 原子数量。其中,五元环C 原子上H 迁移反应的活化能最低,七元环次之,六元环最高。因为不同环状化合物的环张力不同,键角越接近109°28′越稳定。此外,通过对比脂肪链和C 环上的H 迁移反应发现,脂肪链上的H 迁移反应所需能垒均比环上的H迁移反应能垒高。因此,脂肪链上的H 迁移反应比碳环上的H 迁移反应更难。
在整个反应体系中共有六个闭环反应,分别为:CS1→CS2(79.5 kJ/mol)、CS1→CS4(179.6 kJ/mol)、CS7→CS8(135.2 kJ/mol)、CS10→CS11(143.1 kJ/mol)、CS10→CS16(203.6 kJ/mol)、CS13→CS14(266.8 kJ/mol)。虽然同为闭环反应,但反应能垒间存在较大差异,这是由于C 原子的间距离不一,以及C 原子的饱和状况不同引起的。
2.3 对比与验证
将2.2 部分的DFT 计算结果与文献值的活化能进行验证。对C3H3+C4H2加成反应路径的动力学数据进行了比较,结果见表1。使用计算方法分别为CBS-QB3[13,32]、CCSD(T)/cc-pVTZ[33]、B3LYP/6-311G+(3df,2p)[14]。CCSD(T)/cc-pVTZ 是目前精度较高的基组,其主要误差来源是冻核近似,计算精度较高,误差仅为0.06 kcal/mol[34];CBS-QB3 计算误差为1.49 kcal/mol,具有较高的准确性[32];而B3LYP能够在计算精度与成本上获得较好的平衡,饱受大众青睐[34]。因此,以上文献具有一定的代表性。通过对比可以看出,本工作计算结果与文献[13,14,32,33]中的结果一致性较好,证明了本研究的可靠性。通过反应路径计算,获取的动力学参数可用于完善PAHs 的动力学模型。
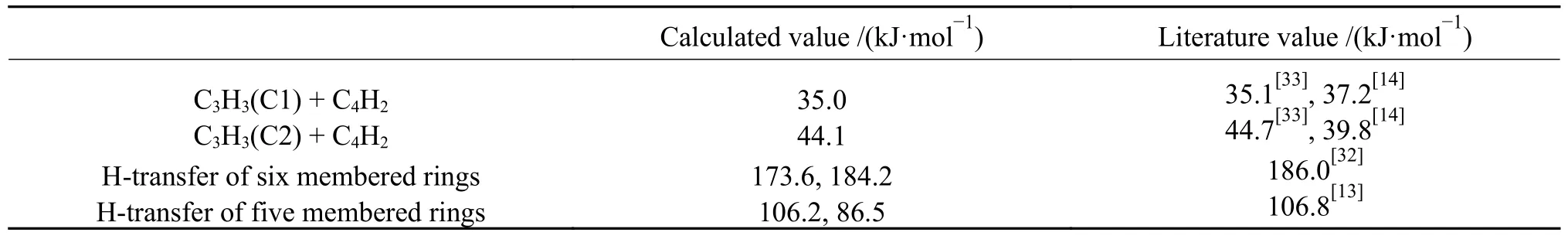
表1 重要反应活化能计算值与文献值Table 1 Calculated values of important reaction activation energy compared with literature values
2.4 基于TST 的反应速率分析
基于DFT 计算结果(自由能、能垒、频率因子0.983、分子的电子能量和转动惯量),利用TST,分别计算500、1500、2500 K 的反应速率常数k、指前因子A、温度指数n、活化能Ea,结果如表2 所示。为了探讨温度对反应速率的影响,通过TST计算300-2500 K,总反应体系中关键基元反应以及限速步的高压极限下(HP)的速率系数,结果如图4 和图5 所示。

表2 由阿伦尼乌斯方程拟合的速率参数Table 2 Rate parameters by fitting the Arrhenius equation

图4 关键反应速率Figure 4 Rate of key elementary reactions

图5 六条路径的反应速率Figure 5 Reaction rate of six paths
图4(a)为两个入口加成反应的速率对比。由图4(a)可知,在300-2500 K,反应C3H3(C1)+C4H2→CS1 的常数明显高于C3H3(C2)+C4H2→CS9。该结果与前文的反应能垒分析结果一致,C3H3(C1) +C4H2→CS1 的反应能垒比C3H3(C2)+C4H2→CS9的反应能垒低8.3 kJ/mol,导致前者更容易进行。随着温度的升高,两者的反应速率逐渐接近。当温度达到2500 K 时,C3H3(C1)+C4H2的反应速率k=4.1 × 109,而C3H3(C2)+C4H2的反应速率k=2.5 × 109。图4(b)为五、六和七元环上的H 迁移反应速率。由图4(b)可知,五元环上的H 迁移反应反应速率(CS2→CS3、CS14→CS15、CS15→CS3)明显高于六、七元环上的H 迁移反应速率。当温度为300 K 时,CS11→CS12 的H 迁移反应速率与CS4→CS5 的反应速率相差不大,随着温度的升高,七元环H 迁移CS11→CS12 的反应速率加快,略高于CS4→CS5 的反应速率。在所有的H 迁移反应中,CS16→CS5 的反应速率最慢。该结果与前文反应能垒的对比结果一致,五元环C 原子上的H 迁移反应速率最快,七元环上H 原子迁移次之,六元环最慢。图4(c)为六个闭环反应的速率常数。由图4(c)可知,在闭环反应中CS1→CS2 的反应速率最快,CS13→CS14的反应速率最慢,在300 K 时反应速率相差35 个数量级。这与反应活化能对比结果一致,CS1→CS2的反应能垒最低为79.5 kJ/mol,而CS13→CS14 的反应能垒最高为266.8 kJ/mol。
根据DFT 计算,速率限制步的动力学速率常数可以代表整个反应过程[31]。图5 为六条反应路径的反应速率常数。由图5 可知,路径1 的反应速率常数明显高于其余五条路径,当温度为300 K时,六条反应路径的反应速率分别为:kpath1=1.7 ×10-2s-1、kpath2=1.7 × 10-31s-1、kpath3=3.4 × 10-49s-1、kpath4=1.9 × 10-25s-1、kpath5=7.4 × 10-37s-1、kpath6=2.5 × 10-51s-1。在低温条件下反应速率的最大差距高达49 个数量级。路径3、5 和6 反应活化能较高,反应速率较慢,在化学动力学上不受支持,反应的发生较为缓慢。随温度的升高,六条反应路径反应速率都会加快,表明,高温条件能够促进反应速率的提升。在整个温度区间,路径2(六元环)和路径6(六元环)的反应速率最慢,路径1(五元环)的反应速率最快,该结果与前文限速步的对比一致,六元环生成的限速步能垒最高,五元环最低。因此,六元环的生成最困难,而五元环生成最具竞争优势。这与Marazana 等[33]在1200 K 实验中产率分析结果相符,六元环的产率最低仅占0.6%-2.7%,而五元环的产率可高达90%以上。
3 结论
本工作利用ESP 和ALIE 预测了较容易发生反应的位点,基于DFT 理论研究了C3H3+C4H2加成反应生成第一碳环的反应路径,详细讨论其反应路径和化学变化。利用TST 理论,计算温度为300-2500 K,各基元反应的动力学参数以及重要中间体的反应速率常数,总结全文可得出以下结论。
在整个反应体系中,涉及到的反应包括加成反应、H 迁移反应和闭环反应三种类型。其中,加成反应克服能垒最低,而H 迁移反应和闭环反应均需较高的活化能,且反应发生缓慢,是决定第一碳环的生长速率的主要反应。
碳环的H 转移能垒和速率取决于C 原子数量。五元环C 原子上H 迁移反应消耗的能垒最低且速率最快,而六元环消耗的能垒最高且速率最慢。与碳环上的H 迁移相比,脂肪链上的H 迁移更难发生。
C3H3与C4H2的加成反应可生成第一个碳环分子的五、六、七元环。其中,五元环CS3 的形成所需能垒最少,反应速率最快,占主导地位。而七元环次之,六元环最少。
猜你喜欢
中学课程辅导·教学研究(2021年8期)2021-07-14
数学年刊A辑(中文版)(2021年1期)2021-06-09
燃料化学学报(2021年5期)2021-06-02
黑龙江电力(2017年1期)2017-05-17
山西大同大学学报(自然科学版)(2016年4期)2016-11-27
环境科技(2016年5期)2016-11-10
新高考·高一物理(2016年3期)2016-05-18
系统工程学报(2015年2期)2015-02-28
电网与清洁能源(2015年2期)2015-02-28
云南中医学院学报(2012年3期)2012-07-31
