
基于全基因组鉴定华石斛优良内参基因
2023-08-14 00:07王雨裴行杰傅钇钧霍恺森梁重钧刘丽岩王佳钮俊
热带作物学报 2023年7期
王雨 裴行杰 傅钇钧 霍恺森 梁重钧 刘丽岩 王佳 钮俊

摘 要:华石斛是海南岛特有的热带兰科植物,具有很高的观赏和药用价值。然而,未确定的内参基因限制了该物种基因表达分析。本研究利用之前华石斛转录组数据,筛选出164 个变异系数低(CV≤0.2)和表达量中等(TPM 在10~300之间)的候选内参基因。为了避免共表达现象对内参基因稳定性分析的影响,根据GO 功能聚类分析结果,从不同的功能群中共筛选出24 个候选基因。为避免模板中可能残留的基因组DNA 在RT-qPCR 扩增过程中产生假阳性,本研究设计了跨越内含子引物,并通过电泳筛选出符合要求的15 个候选内参基因的扩增引物。为了确保RT-qPCR 扩增的特异性,使用LightCycler 96 进行熔解曲线分析。依据15 个候选基因的RT-qPCR 数据,同时综合多个算法分析,在干旱胁迫下,不同软件分别确定CLP1 & SEC23、SEC23 & CPY71、CLP1 & SEC23 和RNG1L & PECT 为最佳内参基因,综合排序后选取SEC23 和CLP1 为华石斛干旱胁迫下最稳定内参基因。在华石斛不同组织中,不同软件分别确定了ADF11& IBR5、PRP19 & CLP1、ADF11 & CLP1、ACBP2 & IBR5 为最佳内参基因,综合分析后确定ADF11 和ACBP2 为不同组织中最稳定内参基因。结合上述干旱胁迫和不同组织分析数据,最终确定IBR5 和CPY71 为综合条件下较稳定的内参基因。内参基因验证表明,与传统的Actin 内参相比,本研究筛选出的内参基因可获得更为精确的RT-qPCR 校准结果,表明所筛选的内参基因具有极高的稳定性。综上所述,本研究为利用转录组数据鉴定稳定内参基因提供思路,首次在华石斛全基因组范围内鉴定稳定内参基因,为进一步研究基因表达和功能奠定基础。
关键词:华石斛;内参基因;转录组;稳定性分析
中图分类号:S682.31 文献标识码:A
华石斛(Dendrobium sinense)是海南岛特有的兰科植物,具有很高的观赏价值和经济价值。
前期的活性成分分析表明,从华石斛中可以分离到菲类、双苄基类、黄酮类和木脂素类等化合物[1-5]。
联苄是石斛属植物的主要酚类成分之一,具有抗肿瘤、抗糖尿病、神经保护等作用[6]。近年来,随着高通量测序技术的快速发展,华石斛公布的基因数量也迅速增加[4]。对探索华石斛花叶发育、胁迫响应和生物活性物质合成途径有关的功能基因和转录因子研究提供了数据基础。然而,华石斛稳定内参基因尚未鉴定,这不可避免地会限制对华石斛基因表达及其功能的探究。
基因表达分析是生物学研究中重要且常用的方法,研究基因表达有助于了解基因的功能[7]。
实时定量聚合酶链反应(RT-qPCR)已经成为目前主流方法,其具有灵敏度高、重复性好、高通量、通用性强等特点[8]。然而,RT-qPCR 产生的相对表达量受许多客观因素的影响,如RNA 的数量、质量和逆转录效率等[9]。RT-qPCR 相对定量时需要稳定内参基因为参考,从而才能获得更准确可靠的表达结果[10]。
目前,管家基因被广泛地用作内参基因进行RT-qPCR 的定量分析[11]。管家基因在不同组织、细胞或实验处理中的表达水平理论上应该是稳定的[11-12],但是许多研究表明,管家基因在不同的实验条件下仍会存在较大的表达波动[13-14]。在不同的物种中,管家基因的稳定性必须经过严格的验证才能作为内参基因进行使用[15]。
随着测序技术的飞速发展,高通量RNA 测序(RNA-seq)已成为非模式物种基因挖掘及功能分析的有力工具。目前,第二代测序(NGS)和单分子实时测序(SMRT)是RNA-seq 最常用的测序技术。2 种RNA-seq 技术被广泛应用于植物的转录组文库构建及差异表达基因识别[4, 16]。通过高通量数据分析,那些在各种组织类型或条件下表现稳定表达水平的基因,可能是潜在的优良内参基因[9, 17]。
基于前期的华石斛转录组数据[4],本研究通过表达矩阵的归一化处理的TPM(transcripts perkilobase of exon model per million mapped reads)算法,计算华石斛文库各个基因的数字表达水平,从中筛选基因表达水平较稳定的潜在候选基因。
通过基因GO 聚类分析,从不同的功能聚类中分别候选内参基因。同时,设计所筛选内参基因的跨内含子引物,利用凝胶成像技术筛选符合条件的内参引物。根据RT-qPCR 数据,利用ΔCt、BestKeeper、NormFinder 和geNorm 四种不同算法软件,评估候选内参基因表达的稳定性并从中确定最佳内参基因。本研究旨在确定华石斛的最佳内参基因,为进一步研究基因表达和功能奠定基础。
1 材料与方法
1.1 材料
野生华石斛采自海南省昌江霸王岭国家级自然保护区。华石斛在25 ℃、相对湿度≥95%的光照培养箱中培养60 d。依据之前的干旱胁迫方法处理植物材料[4],随后收集华石斛根,假鳞茎和叶组织,经液氮速冻并–80 ℃保存。
1.2 方法
1.2.1 转录数据和表达分析 从NCBI Short ReadArchive 数据库中下载转录组数据,华石斛叶片转录组SRR15112264、SRR15112265、SRR15112266,假鳞茎转录组SRR15112267、SRR15112268、SRR15112269,根转录组SRR15112270、SRR1511
2271、SRR15112272。另外SRR14306704、SRR14306705 和SRR14306706 是极度干旱胁迫的华石斛转录组数据(DSC,相对湿度<5%),SRR14306707、SRR14306708 和SRR14306709 为一般干旱胁迫的华石斛转录组数据(DSB,45%≤相对湿度≤ 50% ) SRR14306710 , SRR14306711 和SRR14306712 代表未经干旱胁迫的华石斛转录组数据(DSA,相對湿度≥95%)。根据全长转录组文库[4],用TBtools 软件通过TPM 算法计算基因表达量[18]。
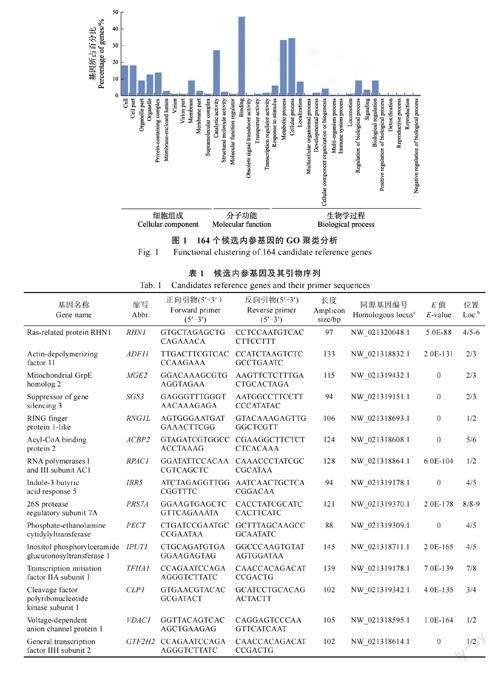
1.2.2 基因筛选与基因聚类 根据RNA-seq 数据的TPM 结果,如果满足变异系数(CV)≤0.2,TPM 水平在10~300 之间,那么就被当作候选基因。为了避免基因共表达可能会影响内参稳定性分析结果,使用Blast2GO 软件对筛选出的候选内参基因进行GO 功能聚类分析[19]。
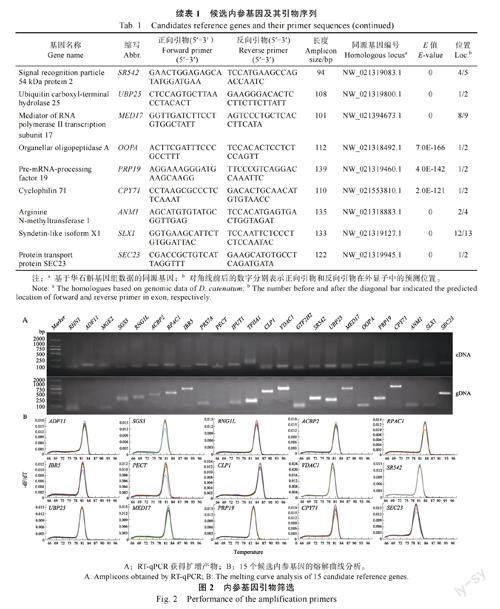
1.2.3 引物设计 华石斛基因组DNA(gDNA)的提取采用植物基因组DNA 提取试剂盒(天根,北京,中国)。总RNA 的提取采用RNA 纯化试剂盒(天根,北京,中国)。Micro-Drop(BIO-DL,上海,中国)检测RNA 纯度和浓度。用反转录试剂盒(Monad,苏州,中国)进行RNA 反转录,RNA 用量2 μg。根据与华石斛同源的铁皮石斛基因组信息( GenBank 登录代码: GCA_001605985.2),利用Primer Quest™ Tool 在不同外显子中设计RT- qPCR 引物。gDNA 和cDNA 的PCR 产物用1.5%琼脂糖凝胶电泳验证。
1.2.4 RT-qPCR 以1∶5 稀释的cDNA作为模板进行RT-qPCR 分析。RT-qPCR 结果使用Light-Cycler 96(Roche, Penzberg, Germany)进行分析。
根据MonAmpTM SYBR®Green qPCR Mix(Monad,广州, 中国)制备20 μL RT-qPCR 反应体系。
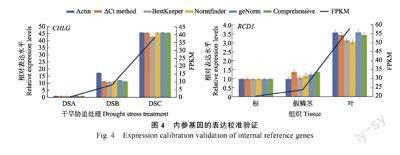
RT-qPCR 扩增从初始步骤95 ℃ 30 s 开始,然后95 ℃变性5 s,55 ℃退火10 s,72 ℃延伸30 s,循环40 次。为了确保扩增的特异性,使用Light-Cycler 96 进行熔解曲线分析。RCD1(Radicalinducedcell death 1)是一种参与多种非生物胁迫反应的应激反应基因[20]。叶绿素合成酶(CHLG)参与叶绿素a 合成的最后步骤之一[21]。本研究利用这2 个基因对所选内参基因的可靠性进行验证。
1.2.5 基因表达稳定性分析 根據E=10slope 公式计算不同内参引物的扩增效率[22]。为了评估内参基因的稳定性,使用RefFinder(http://blooge.cn/RefFinder/)筛选最佳内参基因[23]。这个网站收集了目前主要算法( geNorm[8] 、Normfinder[24] 、BestKeeper[25]和比较ΔCt 方法[26])来对测试的候选内参基因进行稳定性比较和排序。
2 结果与分析
2.1 基于华石斛转录组数据筛选候选内参基因
根据之前的华石斛转录组数据,利用TPM 计算基因表达。最终,共鉴定出164 个符合变异系数(CV≤0.2)和中等表达(TPM 在10~300 之间)的候选基因。GO 聚类分析显示,这些候选基因归属于36 个GO 类别(图1)。为了避免基因共表达对内参基因稳定性分析的影响,因此从不同GO 聚类分支中分别挑选候选基因,共筛选出24个候选内参基因(表1)。例如,RHN1 参与囊泡运输,ADF11 参与肌动蛋白丝结合,MGE2 参与ATP 依赖的蛋白易位,SGS3 参与转录后基因调控,RNG1L 参与蛋白泛素化。
2.2 内参基因引物设计
根据铁皮石斛的基因组数据,对24 个候选基因进行同源性鉴定,提取基因的内含子/外显子结构,设计的RT-qPCR 引物至少跨越一个内含子(表1)。为验证RT-qPCR 引物是否跨内含子,分别以cDNA 和gDNA 为模板进行PCR。cDNA 模板中RNG1、MGE2、RPS7A、IPUT1、TFIIA1、TF2H2、OOPDA、ANM1、SLX1 基因的扩增片段与gDNA模板相似(图2A),说明扩增产物之间不存在内含子,这些候选基因可能会在RT-qPCR 扩增过程中产生假阳性。ADF11 和PECT 的扩增产物在cDNA 模板中达到了预期的大小,而在gDNA 模板中未观察到扩增条带(图2A)。这说明在gDNA模板中,ADF11 和PECT 的内含子过长,无法完成PCR 扩增,因此可以当作候选基因。此外,SGS3、RNG1L、ACBP2、RPAC1、IBR5、CLP1、VDAC1、SR542、UBP25、MED17、PRP19、CPY71、SEC23 以gDNA 为模板的扩增条带较cDNA 模板中扩增条带长(图2A)。这说明在正向引物和反向引物的扩增区域中至少包含一个内含子。综上所述一共鉴定出15 个候选内参基因扩增引物具有跨内含子结构。RT-qPCR 扩增后,这15 个内参基因进行了熔解曲线分析,均呈现单峰(图2B),表明引物特异扩增。因此,选择这15 个基因进行进一步的稳定性分析。
2.3 表达稳定性分析
根据15 个候选基因的RT-qPCR 数据,采用4种工具对其表达稳定性进行评估。基因稳定性分析结果用热图呈现(图3),稳定值越低,颜色越红,说明该基因表达越稳定。在干旱胁迫下,通过比较ΔCt 法、BestKeeper、Normfinder 和geNorm 分析分别确定CLP1 & SEC23、SEC23 &CPY71、CLP1 & SEC23 和RNG1L & PECT 为最佳内参基因,综合排序后选取SEC23 和CLP1 作为干旱胁迫下最稳定内参基因。在华石斛不同组织中,4 种算法分别确定了ADF11 & IBR5、PRP19& CLP1、ADF11 & CLP1、ACBP2 & IBR5 为最佳内参基因,综合分析后确定ADF11 和ACBP2 为不同组织中最稳定内参基因。结合上述干旱胁迫和不同组织分析数据,最终确定IBR5 和CPY71为综合条件下较稳定的内参基因。
2.4 内参基因验证
为验证本研究筛选出内参基因的准确性,以最常见的管家基因Actin 作为内参基因。分别验证了CHLG 在不同组织和RCD1 在干旱胁迫下的表达水平(图4)。与预期的一样,不同内参基因归一化后CHLG 和RCD1 的表达量与转录组测序结果呈现出相似的模式,但存在一些细微的差异。
例如,在不同组织分析中,以Actin 作为内参基因时,CHLG 在根和假鳞茎中的表达量没有显著差异,而以ADF11 和ACBP2 作为内参基因时,CHLG在假鳞茎中的表达量显著上调。后者与转录组数据结果更为一致,这说明本研究筛选出的内参基因可以提供更准确的定量结果。
3 讨论
华石斛是海南岛特有的热带兰科植物,具有很高的观赏和药用价值[1]。该植物附生于海拔1000 m 的山地疏林中树干上,干旱胁迫是其生存中的最主要危害。研究表明华石斛具有丰富的活性药物成分,其生物合成机理已成为研究热点[1-2, 5-6]。
然而,华石斛的稳定内参基因尚未鉴定出来,这限制了相关基因的表达和功能研究。因此,为了推进华石斛基因功能、调控机制、遗传机理等研究,稳定的内参基因鉴定至关重要。
高通量测序技术的快速发展使得非模式物种的基因资源挖掘和表达研究成为可能[4, 9]。此前的华石斛转录组测序为本研究提供了大量的基因数据[4]。将clean reads 映射到基因文庫,通过TPM对基因表达水平进行初步筛选。研究表明内参基因的表达水平过低或过高都会影响表达校准结果[13],
本研究共鉴定出164 个低变异系数(CV≤0.2)和中等表达(TPM 在10~300 之间)的候选内参基因。此外,如果不同内参基因同属一个功能类群,那么很可能这些基因存在共表达现象[27]。为避免共表达对内参基因稳定性分析的潜在影响,候选基因从不同功能簇中挑选,包括一些常规内参基因(如ADF11、PRS7A、CPY71 和UBP25)和不常见基因(如SGS3、IPUT1、VDAC1 和MED17)。
虽然在提取RNA 的过程中,样品经过DNAase处理,去除gDNA 污染,但并不清楚样品中是否存在残留的gDNA。为避免残留gDNA对RT-qPCR分析的影响,本研究按照前人的方法设计了跨越内含子的RT-qPCR 引物[27]。根据凝胶电泳结果显示,9 个候选基因(RNG1、MGE2、RPS7A、IPUT1、TFIIA1、TF2H2、OOPDA、ANM1、SLX1)的扩增引物不符合要求,在后续分析中被剔除。熔解曲线分析表明,剩余的15 个候选基因引物进行特异扩增,可用于稳定性分析。
为了评估这些候选内参基因的表达稳定性,本研究使用了4 种主流算法,包括geNorm[8]、Normfinder[24]、BestKeeper[25]和比较ΔCt 方法[26]。
稳定值最低的候选基因被认为具有最高的稳定性。在geNorm 分析中,所有内参基因的稳定性值均低于阈值1.5[8],说明本研究产生的内参基因的稳定性极高。这一结果可能是由于本研究基于转录组数据对内参基因进行了初步筛选,已将不稳定的基因剔除,与前人对苹果内参基因研究一致[9]。内参基因验证表明,与传统的Actin 内参相比,本研究筛选出的内参基因可获得更为精确的RT-qPCR 校准结果,表明所产生的内参基因具有极高的稳定性。
4 结论
基于华石斛前期的转录组数据,通过广泛的筛选获得理想的RT-qPCR 内参基因。本研究将164 个低变异系数及中等表达的候选基因划分为36 个GO 分类,从中筛选了24 个候选基因。以cDNA 和gDNA 为模板,筛选出了15 个跨越内含子的候选基因引物。综合多种算法分别选取了SEC23 与CLP1、ADF11 与ACBP2、IBR5 与CPY71分别为干旱胁迫、不同组织和综合情况下最稳定的内参基因。本研究首次在华石斛全基因组范围内鉴定了稳定内参基因,为进一步研究基因表达和功能奠定了基础。
参考文献
[1] 蔡彩虹, 谭彩银, 陈惠琴, 王昊, 梅文莉, 宋希强, 戴好富.华石斛化学成分研究(Ⅱ)[J]. 广西植物, 2020, 40(9): 1368-1374.
[2] CHEN X J, MEI W L, CAI C H, GUO Z K, SONG X Q, DAI H F. Four new bibenzyl derivatives from Dendrobium sinense[J]. Phytochemistry Letters, 2014, 9: 107-112.
[3] CHEN X J, MEI W L, ZUO W J, ZENG Y B, GUO Z K, SONG X Q, DAI H F. A new antibacterial phenanthrenequinone from Dendrobium sinense[J]. Journal of Asian Natural Products Research, 2013, 15(1): 67-70.
[4] ZHANG C, CHEN J, HUANG W, SONG X, NIU J. Transcriptomics and metabolomics reveal purine and phenylpropanoid metabolism response to drought stress in Dendrobium sinense, an endemic orchid species in Hainan Island[J]. Front Genet, 2021, 12: 1039.
[5] 谭彩银, 梅文莉, 赵友兴, 黄圣卓, 孔凡栋, 杨宁宁, 宋希强, 戴好富. 华石斛化学成分研究[J]. 热带亚热带植物学报, 2017, 25(2): 189-194.
[6] HE L, SU Q, BAI L, LI M, LIU J, LIU X, ZHANG C,JIANG Z, HE J, SHI J. Recent research progress on natural small molecule bibenzyls and its derivatives in Dendrobium species[J]. European Journal of Medicinal Chemistry, 2020,204: 112530.
[7] VANGUILDER H D, VRANA K E, FREEMAN W M.Twenty-five years of quantitative PCR for gene expression analysis[J]. Biotechniques, 2008, 44(5): 619-626.
[8] VANDESOMPELE J, DE PRETER K, PATTYN F, POPPE B, VAN ROY N, DE PAEPE A, SPELEMAN F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes[J].Genome Biology, 2002, 3(7): 1-12.
[9] ZHOU Z, CONG P, TIAN Y, ZHU Y. Using RNA-seq data to select reference genes for normalizing gene expression in apple roots[J]. PLoS One, 2017, 12(9): e0185288.
[10] DERVEAUX S, VANDESOMPELE J, HELLEMANS J.How to do successful gene expression analysis using real-time PCR[J]. Methods, 2010, 50(4): 227-230.
[11] DHEDA K, HUGGETT J F, BUSTIN S A, JOHNSON M A,ROOK G, ZUMLA A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR[J]. Biotechniques,2004, 37(1): 112-119.
[12] CHEN C M, LU Y L, SIO C P, WU G C, TZOU W S, PAI T W. Gene ontology based housekeeping gene selection for RNA-seq normalization[J]. Methods, 2014, 67(3): 354-363.
[13] THELLIN O, ZORZI W, LAKAYE B, DE BORMAN B,COUMANS B, HENNEN G, GRISAR T, IGOUT A, HEINEN E. Housekeeping genes as internal standards: use and limits[J]. Journal of Biotechnology, 1999, 75(2/3): 291-295.
[14] SUZUKI T, HIGGINS P J, CRAWFORD D R. Control selection for RNA quantitation[J]. Biotechniques, 2000, 29(2):332-337.
[15] LI F, CHENG Y, MA L, LI S, WANG J. Identification of reference genes provides functional insights into meiotic recombination suppressors in Gerbera hybrida[J]. Horticultural Plant Journal, 2022, 8(1): 123-132.
[16] YE J, CHENG S, ZHOU X, CHEN Z, KIM S U, TAN J,ZHENG J, XU F, ZHANG W, LIAO Y. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis[J]. Industrial Crops and Products, 2019, 139: 111547.
[17] CZECHOWSKI T, STITT M, ALTMANN T, UDVARDI M K, SCHEIBLE W R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis[J]. Plant Physiol, 2005, 139(1): 5-17.
[18] CHEN C, CHEN H, ZHANG Y, THOMAS H R, FRANK M H, HE Y, XIA R. TBtools: an integrative toolkit developed for interactive analyses of big biological data[J]. Molecular Plant, 2020, 13(8): 1194-1202.
[19] GÖTZ S, GARCíAGÓMEZ J M, TEROL J, WILLIAMS T D, NAGARAJ S H, NUEDA M J, ROBLES M, TALÓN M, DOPAZO J, CONESA A. High-throughput functional annotation and data mining with the Blast2GO suite[J]. Nucleic Acids Research, 2008, 36(10): 3420-3435.
[20] FUJIBE T, SAJI H, WATAHIKI M K, YAMAMOTO K T.Overexpression of the RADICAL-INDUCED CELL DEAT H1 (RCD1) gene of Arabidopsis causes weak rcd1 phenotype with compromised oxidative-stress responses[J]. Bioscience Biotechnology and Biochemistry, 2006, 70(8): 1827-1831.
[21] SCHMID H C, OSTER U, KÖGEL J, LENZ S, RÜDIGER W. Cloning and characterisation of chlorophyll synthase from Avena sativa[J]. Biological Chemistry, 2001, 382: 903-911.
[22] RAMAKERS C, RUIJTER J M, DEPREZ R H L, MOORMAN A F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data[J]. Neuroscience Letters, 2003, 339(1): 62-66.
[23] XIE F, XIAO P, CHEN D, XU L, ZHANG B. miRDeep-Finder: a miRNA analysis tool for deep sequencing of plant small RNAs[J]. Plant Molecular Biology, 2012, 80(1): 75-84.
[24] ANDERSEN C L, JENSEN J L, ØRNTOFT T F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets[J]. Cancer Research, 2004, 64(15): 5245-5250.
[25] ZHANG K, FAN W, CHEN D, JIANG L, LI Y, YAO Z,YANG Y, QIU D. Selection and validation of reference genes for quantitative gene expression normalization in Taxus spp[J]. Scientific Reports, 2020, 10(1): 1-10.
[26] HOJA-ŁUKOWICZ D, MACIĄŻEK D, KOŚCIELNIAK P,JANIK M E. Innovative GenExpA software for selecting suitable reference genes for reliable normalization of gene expression in melanoma[J]. Scientific Reports, 2022, 12(1): 3331.
[27] NIU J, ZHU B, CAI J, LI P, WANG L, DAI H, QIU L, YU H, HA D, ZHAO H. Selection of reference genes for gene expression studies in Siberian Apricot (Prunus sibirica L.) germplasm using Quantitative Real-Time PCR[J]. PLoS One,2014, 9(8): e103900.
猜你喜欢
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年2期)2017-03-25
中国中药杂志(2017年1期)2017-03-06
中国中药杂志(2016年22期)2017-02-13
价值工程(2016年36期)2017-01-11
中国高新技术企业(2016年32期)2016-12-27
科教导刊·电子版(2016年5期)2016-04-19
计算技术与自动化(2015年3期)2015-12-31
居业(2015年6期)2015-09-05
江苏农业科学(2014年1期)2014-07-18
