
女性肾上腺脑白质营养不良患者的临床和遗传学特征分析
2023-08-15 12:17刘桃桃刘晓黎邬静莹倪瑞隆张梦圆季杜欣
上海医学 2023年6期
刘桃桃 刘晓黎 邬静莹 倪瑞隆 张梦圆 季杜欣 张 梅 曹 立
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是最常见的X连锁隐性遗传的过氧化物酶体疾病,其由ATP结合盒亚家族D成员1(ATP-binding cassette subfamily D member l,ABCD1)基因突变诱发[1-2],发病率约为1∶21 000[3]。ABCD1基因位于Xq28,由9个内含子和10个外显子组成,其编码的肾上腺脑白质营养不良蛋白(adrenoleukodystrophy protein,ALDP)位于过氧化物酶体膜上,主要功能为将酰基化的极长链脂肪酸(very long chain fatty acid, VLCFA)转运至过氧化物酶体内进行β氧化和分解[4]。当ABCD1基因发生突变时,VLCFA不能转运至过氧化物酶体内而特异性地聚积于循环、脑白质、脊髓及肾上腺皮质中[4]。根据起病形式,ALD主要分为脑型肾上腺脑白质营养不良(cerebral adrenoleukodystrophy,CALD)、肾上腺脊髓神经型(adrenomyeloneuropathy,AMN)和女性携带者[5]。临床以CALD最常见,该类型患者多在儿童期起病,临床多表现为认知功能损伤、共济失调、皮质盲和耳聋,其病情进展迅速,多在发病后5~10年内死亡[6]。AMN则是成人患者的常见类型,其以进行性步态痉挛、感觉异常和膀胱功能障碍等脊髓功能异常症状为突出表现,此类患者病情进展缓慢。早期的研究认为,仅有携带ABCD1基因突变的男性患者易出现相关临床表型;近年多项研究[7-9]则发现,女性携带者随着年龄的增长亦可出现神经功能障碍。目前,国内鲜见有关女性ALD患者的临床及遗传学特征的报道,本研究通过分析4例女性ALD患者的临床、影像学、生物化学(简称生化)及遗传学特征,结合文献检索资料,为该疾病的早期发现和及时诊断提供参考。
1 对象与方法
1.1 研究对象 收集并回顾性分析2019年6月—2022年9月在安徽理工大学第一附属医院和上海交通大学医学院附属第六人民医院就诊的4例女性ALD患者的临床及遗传学资料。纳入标准:①基因检测发现携带ABCD1基因致病性杂合突变的女性。②患者有神经系统异常症状,如下肢僵硬无力、步态异常、认知功能减退、精神和行为异常、视力和听力下降、神经性疼痛、感觉异常、括约肌功能障碍,至少包括其中1项。排除标准:排除其他原因引起的上述神经系统的异常。本研究经上海交通大学医学院附属第六人民医院伦理委员会审核并批准(批号:2021-219),所有患者及其家属均签署知情同意书。
1.2 研究方法
1.2.1 资料收集 按照患者就诊的先后顺序分别编号为P1~P4。收集、记录患者的临床资料,包括现病史、既往史及家族史;对患者进行详细的神经系统检查,并记录阳性体征;分析患者的影像学、肌电图、肌肉神经活组织检查(简称活检)、血清VLCFA及肾上腺皮质功能等检查结果。
1.2.2 基因检测 4例患者均进行了基因检测,对于无ALD家族史的患者采用全外显子测序法;对有ALD家族史的患者,采用Sanger测序法验证家族中先证者已存在的ABCD1基因变异位点。
1.3 文献检索 因ALD为相对罕见的遗传性疾病,为佐证本研究收集、分析的4例病例临床资料及基因变异特征,故进一步在中国知网、PubMed中分别以“肾上腺脑白质营养不良”AND“女性”AND“脑白质病”和 “adrenoleukodystrophy” AND “female” AND “leukoencephalopathy”为关键词进行检索,选取、分析有脑白质病变的女性ALD患者的临床、生化、影像学或遗传学的资料及特征。文献资料纳入标准:①基因检测发现携带ABCD1基因致病杂合突变或有ALD家族史的女性;②首发或病程中出现脑部受累的症状,包括精神异常、认知功能障碍,视力、听力异常,癫痫等;③头颅MRI检查显示有脑白质脱髓鞘病灶。文献资料排除标准:排除其他原因引起的神经功能障碍及脑白质异常。

2 结 果
2.1 一般资料 4例患者的发病年龄为(43.75±14.71)岁(范围为23~56岁),病程为(5.75±3.20)年(范围为3~9年)。其中,3例(P2、P3、P4)患者有ALD家族史。患者P1无家族史,在44岁时出现双下肢无力、步态异常,多次至外院就诊,诊断为“腰椎间盘突出、脊髓炎”,给予康复理疗联合糖皮质激素治疗后,症状无改善;起病8年后,至上海交通大学医学院附属第六人民医院就诊,初诊为“遗传性痉挛性截瘫”,行全外显子组测序发现患者携带ABCD1基因致病性杂合突变,故确诊为女性AMN。P2起病后被诊断为“腔隙性脑梗死”;起病2年后,其子出现步态异常,被诊断为“AMN”,患者在进一步的家系共分离验证中被确诊为女性AMN。P3于怀孕后出现双下肢僵硬、无力,多次于外院就诊均未能明确诊断,其子在7岁时因出现认知功能障碍,于外院诊断为“儿童CALD”,至截稿时,其子已9岁,处于植物状态,患者在家系共分离验证中被诊断为女性AMN。P4之子在6岁时出现认知功能减退,于外院诊断为“儿童CALD”,10岁时死亡,患者在家系共分离验证中被确诊携带ABCD1基因变异,该患者在52岁时出现双下肢僵硬、无力、步态异常而被诊断为女性AMN。4例患者的临床特征及基因突变信息见表1。
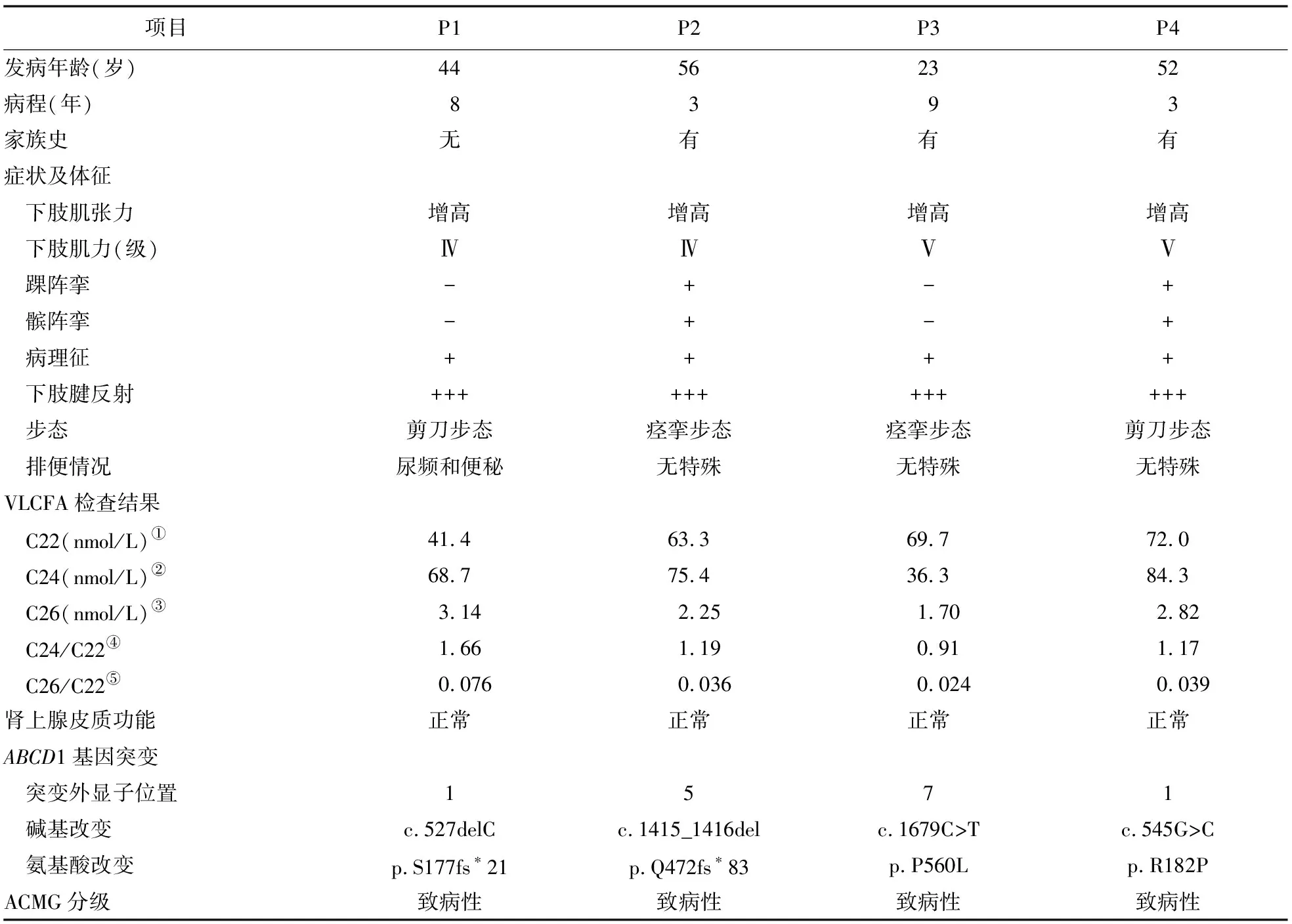
表1 4例女性ALD患者的临床特征及基因突变信息
2.2 临床特征 4例患者均以双下肢僵硬、无力、步态异常为主要表现,不伴有皮肤颜色变黑、头发稀疏。3例患者无明确发病诱因。P3在首次怀孕后出现双下肢僵硬、无力,妊娠结束后症状无缓解;第2次妊娠期间上述症状加重,出现痉挛步态,上下楼梯需手扶栏杆,平地行走速度减慢。病程中,所有患者均无认知功能减退,精神及行为异常;神经科体格检查显示,4例患者均有下肢肌张力增高、腱反射亢进、病理征阳性;其他体征包括:下肢外展肌力下降(P1、P2),踝阵挛、髌阵挛阳性(P2、P4),步态异常(P1、P4为剪刀步态,P2、P3为痉挛步态),高弓足(P1)。同时,P1亦出现尿频、便秘症状。
2.3 生化检查 所有患者血清C22、C24水平均正常,C26水平升高,C26/C22的比值增高。仅P1存在C24/C22的比值增高。4例患者肾上腺皮质功能均正常。
2.4 影像学检查 4例患者均行头颅MRI平扫检查,其中P3、P4分别在发病9年、3年后首次行头颅MRI检查,发现存在脑白质异常,P3表现为双侧额顶叶及脑室周围白质散在、多发的非特异性点状病灶;P4表现为左侧额叶点状,胼胝体压部及双侧侧脑室前、后角对称性片状融合性白质病灶;这2例患者白质病灶均呈T1低信号、Flair高信号。见图1。P1、P3行颈椎MRI检查,P1见多发颈椎椎体黄髓化,各颈椎间盘变性,第4至7颈椎椎间盘轻度突出;P3见颈椎退行性变,第5至7颈椎椎间盘突出,第5至6颈椎椎管狭窄。1例患者(P3)胸椎MRI检查结果无异常。2例患者行腰骶椎MRI检查,P1见第5腰椎至第1骶椎小关节骨质增生,P3见骶椎椎管2~3个囊肿。所有患者均无脊髓病变。

A~C P3头颅MRI检查结果显示双侧额顶叶、脑室周围白质弥漫、散在分布的点状白质病灶(红色箭头所示)。在T1序列上呈低信号(A);在Flair序列上呈高信号(B、C) D~F P4头颅MRI检查显示左侧额叶、胼胝体压部、双侧侧脑室前后角及侧脑室周围白质病灶(红色箭头所示)。在T1序列上呈低信号(D);在Flair序列上呈高信号,即侧脑室周围白质病灶呈双侧点状对称性(E),以及胼胝体及侧脑室前后角病灶呈片状融合性(F)
2.5 其他检查 3例患者行肌电图检查,其中P1双下肢运动及感觉传导速度减慢,波幅减小;P2及P3双下肢运动及感觉传导速度、波幅均正常,仅有双侧F波潜伏期延长。P1行腓肠神经活检,电子显微镜(简称电镜)下可见神经束内大直径有髓神经纤维中度减少,伴再生簇形成,个别有髓神经纤维出现轴索变性,以及轴索和髓鞘间空泡形成,偶见轴索萎缩及薄髓鞘的有髓神经纤维,提示慢性轴索性周围神经病。见图2。

A 有髓神经纤维密度减低,再生簇(提示慢性再生)形成(红色方框所示,×2 000) B 轴索空泡样变性(×8 000)
2.6 基因检测 4例患者均进行ABCD1基因检测(转录本号为NM_00033),结果显示存在4个不同的基因突变,包括2个错义突变[c.545G>C(p.R182P),c.1679C>T(p.P560L)]和2个移码突变[c.527delC(p.S177fs*21),c.1415_1416del(p.Q472fs*83)]。其中,c.545G>C,c.1679C>T和c.1415_1416del均为已报道的致病性变异,c.527delC为本研究首次报道。根据ACMG分级的分类标准[10],该变异被判定为“致病性”突变。
如图3所示,患者P1(Ⅱ-3)存在ABCD1基因移码杂合突变(c.527delC),其2子(Ⅲ-1和Ⅲ-2)均未携带该突变。患者P2(Ⅱ-1)存在ABCD1基因移码杂合突变(c.1415-1416del),其长子(Ⅲ-1)携带该突变,次子(Ⅲ-2)未携带该突变。患者P3(Ⅱ-2)存在ABCD1基因错义杂合突变(c.1679C>T),其长子(Ⅲ-1)携带该突变,并在外院被诊断为“儿童CALD”,无血液标本故未予以验证;其次子(Ⅲ-2)未携带该突变。患者P4(Ⅱ-2)存在ABCD1基因错义杂合突变(c.545G>C),其女(Ⅲ-1)携带该突变,目前无症状;其子(Ⅲ-2)在10岁时因“儿童CALD”死亡。
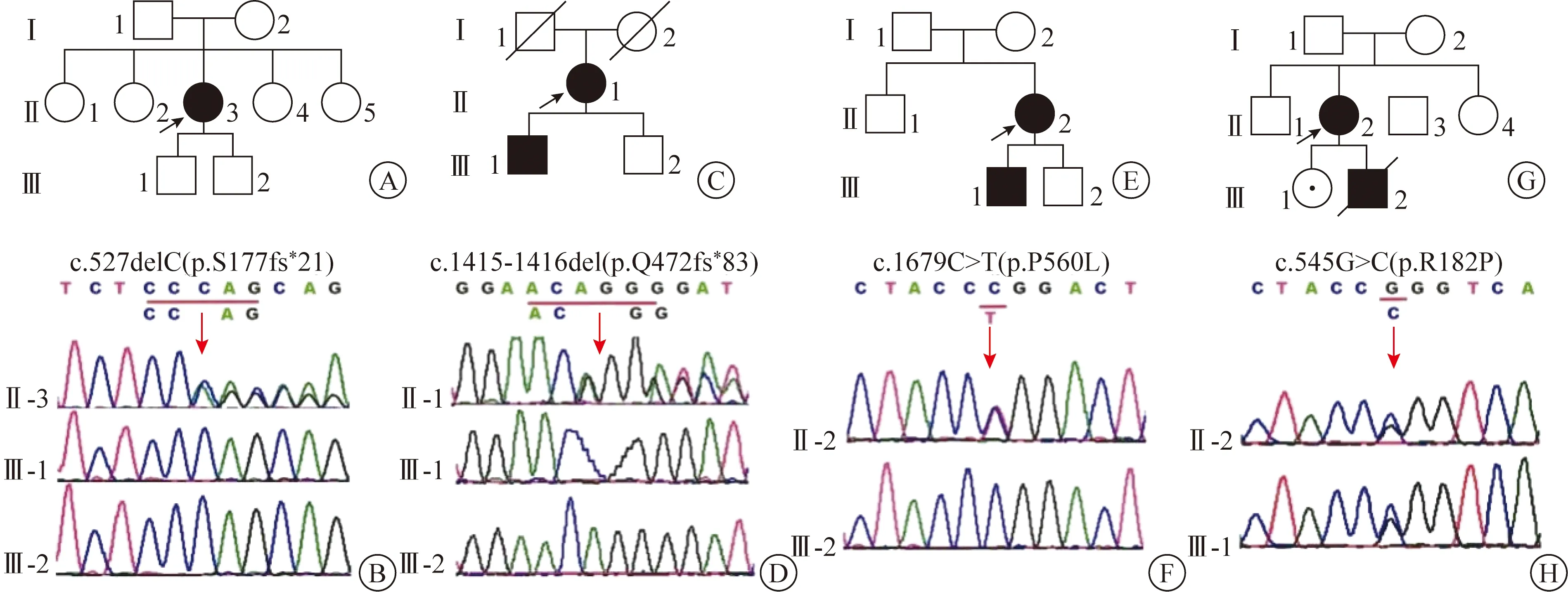
在家系图中,患者以黑色箭头指示,正方形示男性,圆形示女性,黑色实心示患者,圆形加点示无症状携带者,空心示暂无相应临床症状,加斜线表示已死亡。在基因测序图中,ABCD1基因突变位置以红色箭头指示。A、B P1家系图和基因测序图,患者P1(Ⅱ-3)存在ABCD1基因移码杂合突变(c.527delC),其2子(Ⅲ-1和Ⅲ-2)未携带该突变 C、D P2家系图和基因测序图,患者P2(Ⅱ-1)存在ABCD1基因移码杂合突变(c.1415-1416del),其长子(Ⅲ-1)携带该突变,次子(Ⅲ-2)未携带该突变 E、F P3家系图和基因测序图,患者P3(Ⅱ-2)存在ABCD1基因错义杂合突变(c.1679C>T),其长子(Ⅲ-1)携带该突变,其次子(Ⅲ-2)未携带该突变 G、H P4家系图和基因测序图,患者P4(Ⅱ-2)存在ABCD1基因错义杂合突变(c.545G>C),其女(Ⅲ-1)携带该突变,其子(Ⅲ-2)已死亡
2.7 文献检索结果 经过筛选,共纳入26例有脑白质脱髓鞘的女性ALD患者,发病年龄范围为6~79岁,病程范围为4个月~43年(部分未知)。其中,2例患者无家族史,12例有家族史,12例家族史未知。14例患者以记忆力减退,或精神行为异常,或视力减退等CALD样表现为首发症状,7例患者以下肢僵硬、无力,或步态不稳,或括约肌功能异常等AMN样表现为首发症状,1例患者首发表现为肾上腺皮质功能不全,4例患者首发表现未知。12例患者行血液VLCFA检查,结果均显示血液VLCFA水平升高;10例行肾上腺皮质功能检查的患者中,2例结果显示其功能减退。所有患者影像学检查显示,均存在脑白质脱髓鞘病灶,其中9例累及顶和(或)枕叶白质,9例累及脑室周围白质,6例为弥漫性白质病灶,其他患者的白质受累部位包括额叶白质、胼胝体、小脑白质、锥体束、内囊后肢和外侧膝状体。12例患者在文献资料中被记录了ABCD1基因突变信息,其中3例突变位于跨膜结构域,6例突变位于核苷酸结合区域,3例突变位于蛋白质非功能区域。见表2。
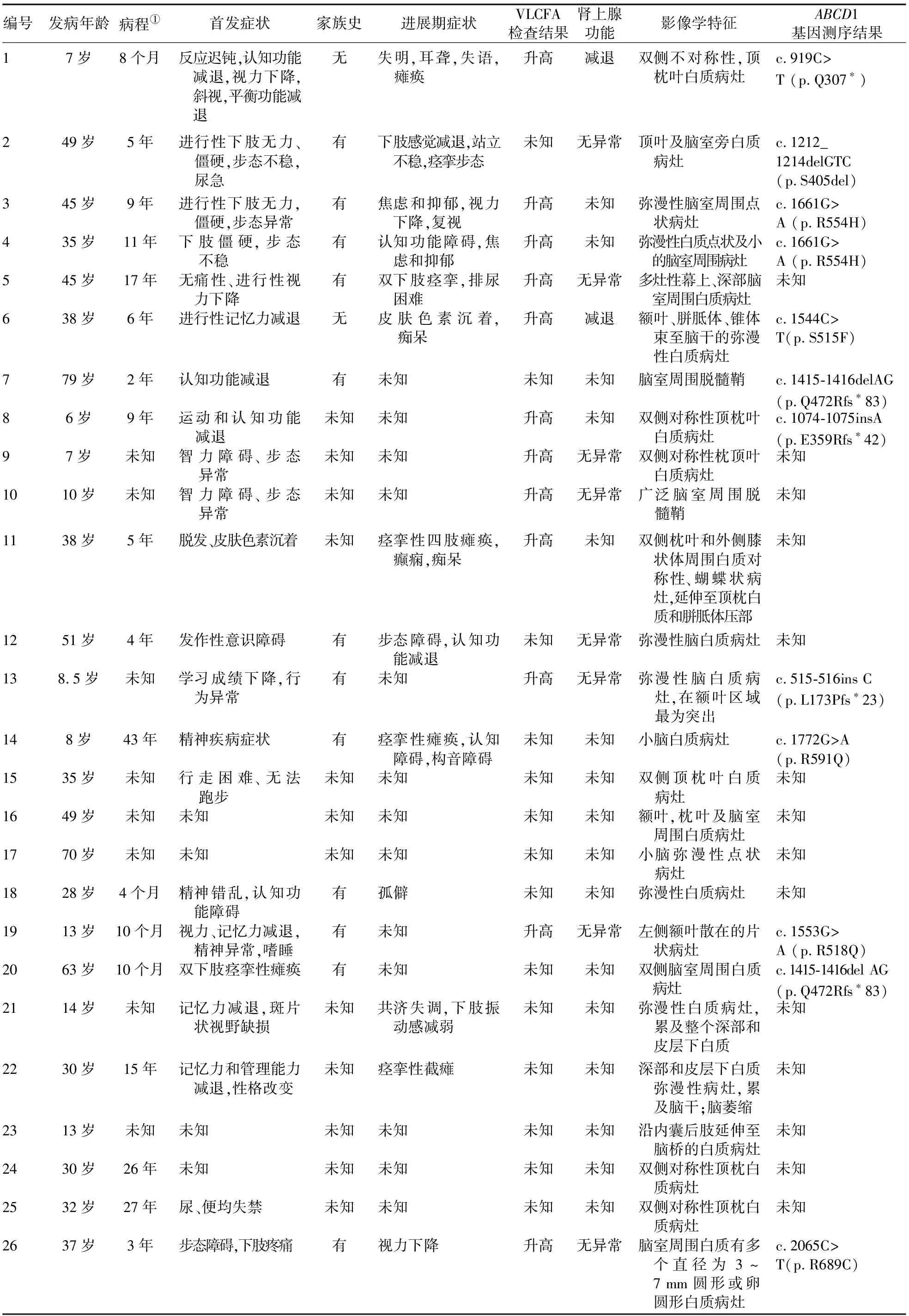
表2 女性ALD患者的临床、生化、影像学特征及基因测序结果
3 讨 论
与多种X连锁隐性遗传疾病一样,早期研究认为携带ABCD1基因突变的女性患者可无明显的临床表现。然而,近期多项研究[9, 11-12]结果表明,女性携带者亦可出现神经系统异常,包括运动功能异常和认知功能障碍等。女性ALD患者常表现为AMN样症状,症状一般较轻且进展缓慢[13],表型外显率呈年龄依赖性[11,14],40岁以下的携带者发病率<20%,而60岁以上的携带者则近90%[11],41岁是表型转换的重要年龄节点[14]。女性携带者进展为CALD的概率<2%[8],患者可表现为精神、行为异常,以及性格改变和认知功能障碍[15]。头颅MRI检查结果的异常可在神经功能异常出现前2年被检测到[16]。在本研究中,P3起病约9年后头颅MRI检查结果显示,双侧额顶叶白质散在、多发的非特异性T1低信号及Flair高信号病灶,P4则在起病约3年后行头颅MRI检查,结果表现为多发性白质脱髓鞘病灶。这2例患者均无明显的认知功能减退及精神、行为异常,P4白质病灶不能明确归因于ALD,因为脑白质信号异常在老年患者中较常见,这些病灶可能缘于局部脑组织的慢性缺血、缺氧[17]。P3为青年女性,无常见的动脉硬化危险因素,白质病变不能用其他病因解释,且这种头颅非特异性的病灶在既往病例中也有报道[15],其是否为脑型早期改变需进一步的随访观察。目前,女性CALD的白质病变模式及临床表现鲜见系统性的研究,故本文总结了既往文献资料中存在脑白质脱髓鞘表现的女性患者的临床及影像学特征,发现儿童及青少年期起病的患者以CALD症状多见,临床表现为学习能力下降、认知功能障碍,以及精神、行为异常;而在成年期出现脑白质脱髓鞘的女性患者中,首发症状复杂多样,患者可表现为CALD的症状,也可以AMN样症状起病,后者在后续的疾病进展中逐渐出现CALD的临床表现及影像学特征,此类患者称为转换型CALD。在疾病早期,白质脱髓鞘病灶最常累及双侧顶枕叶及脑室周围白质,少数患者仅有单侧白质受累,且在原发性CALD与转换型CALD间未见明显差异。最初病灶可呈点状弥漫性或片状融合性分布,随着病情的进展,在疾病的晚期,可表现为弥漫融合性脑白质脱髓鞘。此外,在脑部受累的患者中,患者间的疾病进展情况也存在较大差异,从类似于儿童CALD的快速致死型[12],到缓慢进展性的神经、精神异常[15],患者的生存时间从数月至数十年不等。
目前,女性携带者的发病机制尚不清楚。有研究[18]发现,相较于健康女性,女性携带者易出现X染色体失活(X chromosome inactivation, XCI)偏斜,且更易出现野生型等位基因失活。2002年,Maier等[13]发现XCI偏斜程度与患者神经系统症状严重程度呈正相关。然而,此后Salsano等[18]通过对30例携带者进行分析,未发现有症状的患者与无症状携带者间存在XCI的显著差异。XCI是否能预测ALD携带者的发病尚存在争议[11,13,19]。目前,研究[20]显示,女性AMN的最早发病年龄约为19岁,与本研究中的P3一样,均属于早发型女性AMN,即患者发病年龄较早,可能与遗传、表观遗传、细胞或环境因素有关[7,21]。此外,P3在两次妊娠期间出现步态异常的症状加重,目前仅有少数研究[22-23]报道妊娠可诱发女性携带者起病,妊娠是否及如何诱发女性携带者起病,尚需进一步的研究确证。女性携带者多在家系验证中被发现,对于无家族史的患者,早期诊断困难。P1无家族史,于起病8年后出现剪刀步态,最初被诊断为“遗传性痉挛性截瘫”,行全外显子组测序检测时发现ABCD1基因突变而被确诊。在最初被诊断为遗传性痉挛性截瘫的患者中,约4.8%经基因检测最终修正诊断为ALD[24]。
约57%~80%的女性AMN患者存在周围神经病变[11,25],多数表现为典型的神经性疼痛,部分患者表现为运动诱发的肌肉、骨骼疼痛[25],神经传导检查显示下肢感觉和运动神经动作电位波幅减小,神经传导速度略有减慢,这是轴突损伤和轻度脱髓鞘的典型表现,这些异常可在出现神经系统症状前被检测到[11,26-27]。在本研究中,仅P1出现感觉运动神经传导异常,这可能与本研究纳入的病例数过少,以及患者病程较短有关。此外, P1进行了腓肠神经的活检,其结果与既往研究[28]结果一致,即病灶主要涉及大的有髓纤维,表现为神经纤维密度降低,轴索变性合并髓鞘丢失,未观察到小神经纤维的异常改变。
血清VLCFA水平升高是本病的特征性生化改变,然而约15%的携带者血清VLCFA可维持正常水平[29-30]。在各种类型的ALD患者中,血清VLCFA水平不随时间变化,且与神经症状的发生无显著相关性[14]。因此,临床并不能通过检测VLCFA水平来监测病情进展。在本研究中,4例患者血清VLCFA水平均不同程度升高。与C22及C24不同,C26水平及C26/C22比值不易受饮食及血脂水平的影响[31],是筛查ALD较灵敏且特异的指标。肾上腺皮质功能减退在女性携带者中罕见,其发生率约为1%[32]。本研究中的4例患者肾上腺皮质功能均正常。
ABCD1是ALD唯一的致病基因,编码由745个氨基酸残基组成的ALDP,其中75~352氨基酸残基编码ALDP的跨膜结构域,负责VLCFA的识别和跨膜转运,474~700氨基酸残基编码核苷酸结合区域,能够结合并水解 ATP,为跨膜转运提供能量,这些ALDP功能区是突变的多发部位[30]。本研究中3例患者的基因突变位于功能区内,P2的ABCD1基因c.1415-1416del的AG突变发生在非功能区,该移码突变导致了蛋白质翻译的提前终止,可引起ALDP表达的缺失而致病[30]。在本研究中,携带4种不同基因突变的患者均以AMN样症状起病,病情进展无明显差异,所有患者均无认知功能障碍及精神异常,无肾上腺皮质功能减退,这与既往研究[33]结果相似,即患者病情严重程度与基因突变的位置、类型及ALDP表达量无关,不能通过突变基因来预测病情进展。此外,约5%的突变是由新发突变引起[33]。因此,早期识别及诊断携带者对促进优生优育意义重大。然而,由于女性携带者发病时间晚,且早期临床表现缺乏特异性,因此早期诊断仍面临巨大挑战。
综上所述,携带ABCD1基因突变的女性亦可出现神经系统的异常,症状的发生率随着年龄增长而增高。患者多无肾上腺皮质功能减退,临床症状类似于AMN,以双下肢无力、僵硬和步态异常为突出表现,病情进展缓慢。不足2%的女性携带者存在脑白质脱髓鞘,白质病灶出现在认知及精神异常症状前,多数患者脱髓鞘病灶位于顶枕叶白质及脑室周围白质,个体间病情进展的差异性较大。目前,女性携带者多通过家族史被诊断,对于无家族史的女性患者,早期诊断较为困难。故对临床上疑诊为单纯型痉挛性截瘫的女性患者,应注意与AMN的鉴别,尽早行ABCD1基因检测可明确诊断。
猜你喜欢
保健医苑(2023年2期)2023-03-15
现代临床医学(2022年4期)2022-09-29
中老年保健(2021年12期)2021-08-24
医学与法学(2020年3期)2020-09-18
电子制作(2018年18期)2018-11-14
自动化学报(2018年6期)2018-07-23
故事作文·高年级(2017年3期)2017-04-12
河北医学(2016年5期)2016-12-01
实用临床医学(2016年8期)2016-06-07
中华老年多器官疾病杂志(2016年8期)2016-05-14
