
基于BSA重测序的辣椒CMS恢复基因连锁分子标记开发
2023-09-20 11:23王萌赵虎徐晓美潘尧铧赵曾菁吴星王日升
热带作物学报 2023年8期
王萌 赵虎 徐晓美 潘尧铧 赵曾菁 吴星 王日升

关键词:辣椒;全基因组重测序;胞质雄性不育;恢复基因;分子标记
辣椒(Capsicumspp.)是世界最大的调味料作物和世界第三大蔬菜作物,也是我国种植面积最大、加工方式最多、消费功能最多的蔬菜和最大的调味品[1]。目前辣椒杂交制种生产仍较多采用人工去雄,种子生产成本高,与国外品种相比缺乏市场竞争力[2]。利用辣椒胞质雄性不育(CMS)进行三系杂交制种不仅可以确保杂交种子纯度,更有利于保护品种的知识产权,是辣椒育种发展的主要趋势[3]。开发与辣椒CMS恢复基因紧密连锁的分子标记,大规模对自交系及中间材料进行恢复基因筛选,可大大提高三系育种效率。
辣椒CMS育性恢复的遗传控制多样且复杂,多数研究者认为辣椒Rf基因由1个显性基因控制的[4-5],WEI等[6]认为辣椒CMS恢复基因(Rf)是受2个主加性-显性上位基因和1个加性-显性多基因控制,也有认为辣椒CMS育性恢复与1个主QTL和4个小QTL有关[7]。针对辣椒Rf基因,前人已经开发了多种类型的分子标记用于辅助育种,这些标记包括随机扩增多态性(RAPD)标记[8]、简单重复序列(SSR)标记[9]、插入/缺失(InDel)标记[10]、切割扩增多态性序列(CAPS)标记[11]、序列特征扩增区(SCAR)标记[12]和竞争性等位基因特异性PCR(KASP)标记[6,13]等。其中应用最广泛的标记CRF-SCAR[12,14]在不同自然群体中对育性恢复性状的准确率最高,同一標记在不同群体间准确率差异较大,准确率较高的报道有89.1%[4]、79.2%[15]和100%[16]。这些结果进一步说明,辣椒基因组包含多个候选Rf基因,不同恢复系可能具有基因型特异性的Rf基因[11,15,17-18],目前尚无通用标记,特异的恢复基因需要开发相应的标记才更有效。
目前,最广泛使用的CRF-SCAR标记[15,17]不能区分本单位的不育系014A和恢复系014C,说明恢复系014C可能具有不同Rf基因,需要开发相应的连锁标记。深度测序结合BSA法,在不构建遗传图谱的情况下,可快速定位正向遗传学的性状位点[19],快速筛选目标基因以获得紧密连锁分子标记[20],目前已用于多种作物质量性状或主效基因的定位,如尹明智等[21]利用该方法定位了油菜野芥胞质雄性不育恢复基因;LI等[22]利用BSA法结合基因组测序和转录组测序,联合分析获得了大白菜中与叶状头形成相关的共同候选基因。本研究以不育系014A和恢复系014C构建了F2分离群体,利用BSA法结合全基因组重测序,获得辣椒CMS恢复基因相关定位区间,根据区间内的差异SNP/InDel设计引物,筛选恢复基因连锁分子标记,为加速选育辣椒CMS恢复系奠定基础。
1材料与方法
1.1材料
以不育系014A×恢复系014C构建F2分离群体,正常田间管理,于花期调查育性,构建测序所需基因可育混池和不育混池,可育混池单株分别隔离,单株留种,每个单株种植30个子代,调查育性分离情况,判断可育混池内单株基因型为RfRf或Rfrf。试验材料种植于广西农业科学院基地。
1.2方法
1.2.1育性调查2020年对F2群体1008个单株进行插牌编号,田间正常管理,于辣椒开花期开展3次以上育性调查并记录,参考花粉指数(PI)法:根据肉眼观察花粉数量分为1~4级,每株调查10朵花,在开花当天进行观察。1级:花药上布满花粉,同可育亲本无明显差异;2级:有花粉,但不及可育亲本的一半,花粉量明显减少;3级:有很少量的花粉;4级:花药干瘪皱缩,无可见的花粉。对于不易判定等级的调查的单株,采用多次调查的方法,同时调查自然坐果率和果内种子数量进行辅助判断并记录。
1.2.2建池、测序与数据处理在F2群体采集单株叶片提取DNA,同时分别选择1级和4级各30个单株,分别取幼嫩叶片0.1g用于DNA的提取,DNA分别等量混合构建不育基因池和可育基因池。2个混池连同亲本建库后使用HiseqX10PE150上机测序,测序深度为30×。样本由广州基迪奥生物科技有限公司完成建库和测序。测序得到的原始数据先进行过滤,获得cleanreads,再利用Burrows-WheelerAligner(BWA,v0.7.16a-r1181)将过滤后的reads与辣椒参考基因组Ensembl_release47(http://plants.ensembl.org/Capsicum_annuum/Info/Index)进行比对。比对结果使用GATK(v3.5)VariantFiltration模块对SNP和InDel进行变异检测。
1.2.3基于SNP-index的BSA分析与Fisher精确检验对辣椒育性连锁定位区间进行筛选时,首先过滤不育和可育混池中SNP-index均小于0.3或大于0.7的位点,计算各混池的SNP-index和混池间的Δ(SNP-index),然后以滑动窗口法对Δ(SNP-index)在各个染色体上的分布制图。置信区间设置为95%和99%,取正向置信水平99%以上窗口作为候选区间[23]。
采用Fisher精确检验法(SPSS21.0)对2个混池中的等位基因深度比例进行检验,显著性使用P值表示,再次按照滑窗的方法对计算获得的P值结果进行拟合,取P值的-log10后绘制曼哈顿图。原始的P值进行FDR校正后获得q值,筛选q值小于阈值(0.05)的位点作为显著位点,连续的显著位点合并成一个区间,获得显著区间。
1.2.4DNA提取与引物筛选采用改良的CTAB法提取亲本及F2群体所有单株DNA,检测合格后将浓度统一调整至50ng/μL用于后续试验。根据定位区间获得的基因、InDel/SNPs信息,结合前人报道恢复基因类型信息,每个基因至少设计1对以上的引物,优先选择位于外显子区域的SNP和多态性差异5bp以上的InDel作为第一轮SSR/InDel分子标记,筛选在亲本间呈现多态性的引物。在第一轮多态性引物附近,第二轮根据基因信息和InDel/SNPs设计更多的标记,再次利用双亲进行标记筛选,然后结合F2、F3育性田间调查结果,利用确定了基因型的30个可育混池单株(基因型为RfRf或Rfrf)、30个不育混池单株(基因型为rfrf)进行筛选,获得准确率最高的分子标记,最后使用F2群体进行准确率验证。
提取目标位点两翼各150bp的碱基序列,使用Oligo6软件进行引物设计。引物序列均由生工生物工程(上海)股份有限公司合成。PCR反应总体系均为10μL,其中50ng/μL模版DNA1μL,TaqDNA聚合酶5μL,上下游引物(10μmol/L)各0.5μL,ddH2O3μL。PCR扩增程序参照王日勇等[24]的方法。扩增产物用8%变性聚丙烯酰胺凝胶110V电泳分离95min,用银染法进行显影;根据标记特征,分离群体内验证也可使用2%琼脂糖,电压120V,电泳35min;观察结果并拍照保存。
2结果与分析
2.1F2代分离群体育性调查
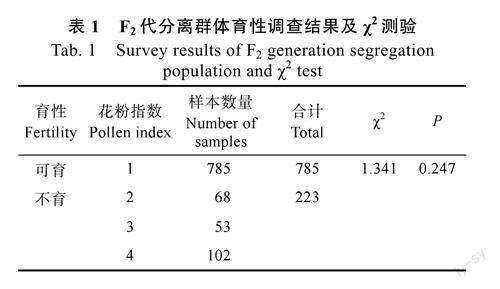
通过不育系014A和恢复系014C杂交构建的F2代分离群体1008个单株育性调查结果表明,花粉正常可育单株为785株,不育的单株为223株,可育、不育分离比例均接近3∶1,按1对基因的控制模式分别进行χ2测验,χ2值为1.341,P值为0.247,P>0.05,符合理论预测,即可育和不育分离比符合3∶1的分离规律(表1),表明辣椒CMS育性恢复性状受1对显性基因控制。30个测序用的极端可育单株通过种植调查F3群体分离情况,育性不发生分离则亲本为纯合可育(RfRf),育性发生分离则亲本为杂合可育(Rfrf),连同30个纯合不育单株(rfrf)用于后续分子标记筛选与验证。
2.2重测序混池数据质量评估
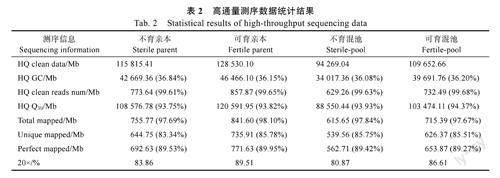
根据亲本和混池重测序的相关数据结果显示(表2),此次测序共得到450.60Gb原始数据,经数据质控过滤后获得448.27Gb高质量有效数据。样品的GC碱基含量为36.08%~36.84%,测序质量控制标准Q30>93.75%。不育、可育测试样品与参考基因组的比对率分别为97.84%、97.67%。基因组覆盖度20×比例约为80%以上。全基因组范围内分别检测到950253个InDel和15142397个SNP。由此可知,本研究样本数据量足够,GC分布正常,测序质量合格,测序数据与参考基因组比对结果正常,覆盖度饱和,可用于后续的变异分析及目标性状的基因定位。
2.3与辣椒CMS育性关联的连锁区间定位
基于SNP-index的BSA分析结果显示,正向置信水平99%以上窗口有12个区间分别位于6号、8号、11号染色体上(表3,图1)。由图1可知,6号染色体定位区间峰值高、峰形宽大,8号染色体存在多个小峰,11号染色体也存在一个峰值高的狭窄峰。由表3可知,6号染色体的第一个定位区间最大,长度为26.72Mb,约占总区间长度32%,其中包含540个基因,约占基因总数的64%,基因分布相对集中。
为进一步缩小定位区间,采用Fisher精确检验法对2个混池中的等位基因深度比例进行检验,得到基于-log10(p)值的全基因组分布曼哈顿图(图2),获得的定位区间为6号染色体1.44~8.28Mb,该区间内包含4441个SNP、266个Indel和227个基因。
2.4分子标记开发
根据6号染色体1.44~8.28Mb候选区间内227个基因及SNP/InDel位点,共设计了316对引物,扩增亲本,筛选出27对在亲本间差异显著的标记。根据基因注释获得的3个恢复基因候选基因,然后重点在这3个基因附近设计引物并筛选,最终获得了能稳定扩增出特异条带的标记OP59和PP5。OP59引物序列为F:5-TGGAACAGAGTCATATTTTTCTTTCAT-3、R:5-CCAATTCCGATAAAGGGTTTT-3。PP5引物序列为F:5-TCATTCTTTAGGGGAAGCTTAGG-3、R:5-CGGTGTGGACAGACATTTCA-3。根据SNP位点设计的标记OP59,在纯合可育材料可扩增出282bp条带,不育材料可扩增出278bp条带,杂合单株能扩增出2条带(图3)。根据InDel位点设计的标记PP5,在不育亲本、F2杂合可育单株、不育单株中均可扩增出约300bp的条带,而纯合可育亲本和F2纯合可育单株中无扩增条带,该标记可使用琼脂糖电泳更方便快速(图4)。2个标记在极端群体验证中准确率均达到100%。
将这2个标记在F2代分离群体随机挑选的456个单株进行验证,结果表明,2个标记在重测序的极端群体中准确率均达到100%。共显性标记OP59在不育的群体中准确率为100%,在可育单株中准确率为97.21%。標记PP5在不育单株和纯合可育单株中准确率均为100%。根据与参考基因组比对结果,标记OP59根据位于6号染色体3877192bp处SNP位点设计,参考碱基为A,突变碱基为G,该位点位于恢复基因候选PPR基因T459-15819基因间区,距离为17232bp;标记PP5根据位于6号染色体3897138bp处InDel位点设计,参考碱基为G,突变碱基为GT,该位点位于该基因下游318bp。
3讨论
本研究表明,辣椒CMS核内恢复基因经BSA结合法结合全基因组重测序,比对参考基因组Ensembl_release47,经Fisher精确检验,定位在6号染色体顶端1.44~8.28Mb区间,区间长度6.84Mb。WEI等[6]通过基于转录组测序的BSR-seq方法,通过与参考基因组Zunla-1比对,将恢复基因定位在6号染色体末端16.8Mb的区间,其使用的试验材料包含的恢复基因被认为是受2个主加性-显性上位基因和一个加性-显性多基因控制。ZHANG等[25]也是使用BSA结合基因组重测序的方法在6号染色体的两端分别获得了一个恢复基因,该材料中辣椒CMS育性恢复同时受2对基因控制。本研究所使用的试验材料F2群体中可育∶不育符合3∶1,因此育性恢复受1对基因控制,这与WEI等[6]、ZHANG等[25]的试验材料不同,推测确实含有不同的恢复基因。
在许多农作物中多个雄性不育恢复基因(Rf)已经被鉴定,大多数恢复基因属于PPR基因(编码蛋白含有五肽重复序列),如水稻[26]、油菜[27]、棉花[28]等。在辣椒作物上,前人报道的恢复基因有PPR6[11]、PPR46[11]、NEDD8[18]、长链非编码RNA[29]等。尹明智等[21]通过对基因定位的候选区域进行序列分析和基因注释,发现其中的PPR基因,再进行基因克隆及功能验证,这将是分析恢复基因的一种有效手段。本研究初步获得的候选基因T459-15819也是属于PPR基因,该基因是否为调控辣椒育性恢复的关键基因,以及如何影响育性的恢复还需进一步分析验证。
本研究获得的标记OP59属于共显性标记,不仅能够区分基因纯合可育(RfRf)植株和不育(rfrf)植株,还能鉴定出杂合可育(Rfrf)的植株,且准确率高,聚丙烯酰胺凝胶电泳即可分辨,在实际应用中非常简便有效。标记PP5琼脂糖凝胶电泳检测即可,实际应用中可先用PP5检出纯合可育株,然后再用OP59检出其他类型。
本研究构建的F2群体中,根据花粉指数法将单株划分为不同的等级,花粉量减少的2、3、4等级全部归为不育,经χ2测验,可育和不育分离比符合3∶1,推测辣椒CMS育性恢复性状受1对显性基因控制,这与多数研究结果一致[4-5,9,14]。从本研究不育群体实际调查数据来看,单株间花粉量存在一定的差异,因此,本研究使用的恢复基因除受1对显性基因控制外,在6号、8号、11号染色体上也可能存在育性修饰的微效基因,或基因表达受到环境影响,仍需进一步研究。
猜你喜欢
少儿科学周刊·少年版(2022年18期)2022-05-30
少儿科学周刊·少年版(2022年18期)2022-05-30
少儿科学周刊·少年版(2022年18期)2022-05-30
小哥白尼(神奇星球)(2021年6期)2021-07-28
中外文摘(2020年9期)2020-06-01
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
山东农业科学(2016年12期)2017-01-21
天津农业科学(2016年12期)2017-01-11
