
蛋白质的圆二色光谱测量中圆二色值不确定度评估
——以细胞色素C为例
2023-10-09 10:31严定策武利庆
光谱学与光谱分析 2023年10期
程 红,严定策*,武利庆,徐 俊
1. 华中科技大学分析测试中心,湖北 武汉 430074 2. 中国计量科学研究院前沿计量科学中心,北京 100029 3. 华中科技大学能源与动力工程学院,湖北 武汉 430074
引 言
在自然界中,手性是生物分子如蛋白质、氨基酸、碳水化合物、核酸等本身固有的属性[1-2]。圆二色(circular dichroism,CD)光谱作为一项用于研究手性分子的光谱技术,在这些物质的研究中被广泛应用,其中以蛋白质尤为突出。通过CD光谱学对蛋白质进行分析,可以鉴定和评估其二级结构[3-4],检测细微或显著的构象变化,研究蛋白质动态变化和/或配体结合效应等[5-6],以获得关于所研究蛋白质的快速和全局结构信息,因此CD光谱学被世界各地的研究人员用作结晶学、核磁共振和分子动力学的补充技术[7]。
在CD光谱中,远紫外区域(通常为180~260 nm)的光谱可以给出蛋白质二级结构含量的总体指示,因为该区域的主要生色团是分子的肽主链。在蛋白质的规则二级结构中,肽键是高度有规律排列的,排列的方向性决定了肽键能级跃迁的分裂情况[8]。对远紫外CD光谱进行解卷积,可以提供二级结构组成的定量估计,但普通生物制药缓冲液和赋形剂可能会产生干扰,糖基化、聚糖缀合、其他翻译后变化或异常氨基酸组成也会影响[9]。用CD方法获得的结构信息有时会受到原始CD数据质量的影响。正如Miles和Wallace所述,在任意两台仪器中进行的CD测量,即使在可比较的条件下,相同样品的光谱幅度(elipticity椭圆度,CD scale,也称圆二色值)或波长也可能存在细微差异。光源、最终的光度输出和各个仪器之间入射光偏振的微小差异是导致圆二色值或波长产生差异的可能原因。为了避免这些差异导致数据的不可比性,在采集CD数据之前,必须对CD仪器进行校准,包括圆二色值和波长,因为CD解卷积分析强烈依赖于圆二色值。
最近,人们认识到蛋白质的高阶结构要得到更好的表征,必须重视这些数据的客观比较[10-11]。要输出统计上有效的数据,就需要清楚地了解光谱不确定性的来源、如何纠正它们以及其对结果有效性的影响。Maurice等[12]采用一次国际比对的数据利用摩尔椭圆度讨论了蛋白质的圆二色光谱谱图的不确定度;Jones[13]比较了一个公共数据库(protein circular dichroism data bank,PCDDB)中108个d-10-樟脑磺酸的校准数据,对这些光谱位于290和190 nm附近的峰进行了统计评估,发现仪器之间峰波长的平均值差异显著;Victor等[14]采用蒙特卡罗模型(Monte Carlo-like model)评估了使用CDSSTR、Selcon 3和ContinLL算法对肌红蛋白的理论CD谱进行二级结构预测的不确定度。本工作以一种α-螺旋为主导的蛋白质—细胞色素C为实验对象,考察了圆二色光谱测定蛋白质实际测试过程中圆二色值θ的不确定度。圆二色光谱是为一段波长下椭圆度θ的测量值,通常θ的单位用mdeg表示。一般情况下,θ值和CD吸光度的差值(ΔA)不在实验室之间或实验室内进行比较[12],但是本实验中考虑所用比色皿的光程长度、仪器的校准状态和溶液的浓度,期望通过特定蛋白质在特定浓度下的θ值不确定度的评估,找出影响测量结果的主要因素,可以设法消除或降低这些因素的影响,改进测量方法,提高CD数据的可比性和可靠性,为进一步进行实验室间比对提供参考。
1 实验部分
1.1 仪器设备
日本Jasco公司J-810型圆二色光谱仪;波兰RADWAG公司AS 60/220 R2型分析天平(经湖北省计量测试技术研究院检定合格,在检定合格有效期内使用);法国Millipore公司Milli-QAcademic型纯水机(经湖北省计量测试技术研究院检定合格,在检定合格有效期内使用);德国Eppendorf公司Researchplus单道可调式移液器,20~200 μL,100~1 000 μL(经湖北省计量测试技术研究院检定合格,在检定合格有效期内使用);光程长为10 mm的石英比色皿1支、光程长为1 mm的石英比色皿3支 (Starna Scientific Ltd. Hainault,Essex IG6 3UT,UK);10 mL容量瓶一支(经湖北省计量测试技术研究院检定合格,在检定合格有效期内使用)。
1.2 样品
d-10-樟脑磺酸铵(ACS,购自日本Jasco公司,部件号:0730-0358,纯度99%);细胞色素C(5 mg,棕色玻璃瓶装,购自中国国家标准物质资源共享平台,中国计量科学研究院研制二级标准物质,GBW(E)100153。在样品有效期内使用。)实验用水为超纯水(电阻率为18 MΩ·cm)。
1.3 方法
1.3.1 溶液的配制
精确称取樟脑磺酸铵样品6.77 mg,使用移液器精密加入1.13 mL超纯水,完全溶解后,使用移液器精密移取1 mL,置10 mL容量瓶,使用超纯水定容至刻度线,浓度为0.6 mg·mL-1。
在细胞色素C样品瓶中精密加入超纯水1 mL,完全溶解后精密移取100 μL至离心管,再精密加入900 μL超纯水稀释,摇匀,精密移取该溶液400 μL至离心管,再精密加入3 600 μL超纯水,摇匀,浓度为0.05 mg·mL-1,平行配制两次。
将所有样品溶液放置在冰箱(4 ℃)中。在进行CD测量之前,让溶液在此温度下平衡至少一整夜。
1.3.2 仪器校准
从冰箱中取出樟脑磺酸铵样品溶液,在室温[(23±2)℃]下平衡1 h,并在进行CD测量之前将样品装入光程长为10 mm的石英比色皿内,根据如下参数测量样品的CD光谱:
扫描范围:200~400 nm
累积次数:1次
响应时间:1 s
带宽:1 nm
灵敏度:Standard(100 mdeg)
步长:1 nm
扫描速度:50 nm·min-1
根据樟脑磺酸铵溶液的出峰情况调整仪器,使291 nm附近谱峰的圆二色值为(190.4±1) mdeg。记录6个重复光谱。
1.3.3 样品溶液的测试
在完成仪器校准当天,将细胞色素C样品溶液从冰箱中取出,在室温[(23±2)℃]下平衡1小时。在进行CD测量之前,先测试溶剂空白-超纯水在光程长为1 mm的石英比色皿的圆二色光谱图,然后将样品装入同一个比色皿内进行CD测量,测试中保持样品测试与空白测试时比色皿的方向一致。根据如下参数测量样品的远紫外CD光谱。记录样品的6个重复光谱(连续测定,重复测量不重新装样)。2个平行溶液分别使用3支比色皿测试,共测试36次。
扫描范围:190~260 nm
累积次数:6次
响应时间:1 s
带宽:1 nm
灵敏度:Standard(100 mdeg)
步长:1 nm
扫描速度:50 nm·min-1
1.4 测试结果
1.4.1 仪器校准结果
根据291 nm附近的谱峰圆二色值的情况调整后,重复六次的结果见表1。

表1 0.6 mg·mL-1樟脑磺酸铵水溶液的圆二色值Table 1 CD magnitude of ammonium d-10-camphorsulfonate (ACS) with a concentration of 0.6 mg·mL-1 in pure water
由于仪器测得的圆二色值不是绝对值,本文只考虑这些值的可比性的实际要求。虽然我们将CD仪器使用樟脑磺酸铵校准到参考CD值(190.4±1) mdeg @(290±1) nm,但这个赋值没有相关不确定度,且不可溯源。CD测量的可溯源性至今仍是一个挑战,不在本文讨论的范围。此外,即使是在参考波长291 nm对仪器进行了充分校准,也无法断定在所有波长下的响应都是相同的,因此受到当前方法的限制,假设仪器响应曲线为平直线,即响应值与波长无关。在以下建立的不确定度模型也是基于这样的假设。
1.4.2 细胞色素C的测试结果
对2个平行样品溶液使用3支光程长1 mm比色皿装样,每个重复6次测量,总计测量36次,得到的CD谱图经过平滑处理后,以250~260 nm附近的平直谱线为基线,记录谱图中222 nm附近的峰高值,即为圆二色值,见表2,单位用mdeg表示,数据精确到小数点后两位。

表2 0.05 mg·mL-1细胞色素C水溶液的圆二色值Table 2 CD magnitude of Cytochrome C with a concentration of 0.05 mg·mL-1 in pure water
2 结果与讨论
2.1 模型的建立
光学活性物质对于左旋圆偏振光和右旋圆偏振光的吸收是不同的,吸光度的差值与吸光度同样符合Beer-Lambert定律。
ΔA=c×l×Δε
(1)
式(1)中,ΔA为左、右吸光度值的差值;c为溶液浓度,单位为mol·dm-3;l为比色皿光程长,单位为cm;Δε为左、右摩尔吸光系数的差值,单位为mol-1·dm3·cm-1。
仪器给出的原值即圆二色值,是光学活性物质吸收左、右圆偏振光后形成的椭圆偏振光的椭圆度,与吸光度差值的关系为
θ=32.982×ΔA
(2)
联合式(1)和式(2),可以得出式(3)
θ=32.982×c×l×Δε
(3)
θ的单位为毫度(millidegree,缩写为mdeg),是一种非标准的平面角单位,被广泛用于CD光谱学领域[15]。1 mdeg等于10-3度或约17.4 μrad。
2.2 不确定度来源分析
根据式(3)中的函数关系,圆二色值的不确定度来源主要有以下几个方面:测量重复性;样品溶液浓度;比色皿光程长;左、右摩尔吸光系数的差值。2.3节对以上几个方面的不确定度进行计算。
2.3 A类不确定度评定
与CD相关测量的不确定度很大程度来源于仪器本身,潜在的噪声源包括光源、光电倍增管、光电调制器、锁定放大器等,这些与仪器性能相关的标准不确定度可以通过多次重复测试使用统计学方法进行评估。环境温、湿度的影响也包含在重复性测量中,下述不再考虑该因素。
对表2中的数据进行统计分析,36次平行测试0.05 mg·mL-1的细胞色素C的圆二色值平均值为-4.527 mdeg,A类标准不确定度为
由重复性引入的相对标准不确定度为
2.4 B类不确定度评定
2.4.1 蛋白溶液浓度的不确定度
细胞色素C样品溶液浓度的不确定度由细胞色素C的含量不确定度以及配置储备液及稀释过程引入,以下分别进行计算。
(1) 查细胞色素C标准物质证书,相对分子量标准值为12 355,扩展不确定度为16,通过相对分子量推算得其纯度的相对标准不确定度为
(2) 溶液配置过程为:在细胞色素C样品瓶中精密加入超纯水1 mL,完全溶解后精密移取100 μL至离心管,再精密加入900 μL超纯水稀释,摇匀,精密移取该溶液400 μL至离心管,再精密加入3 600 μL超纯水,最终浓度0.05 mg·mL-1。查单道可调式移液器检定证书,得1 000 μL容量允差为±1.0%,200 μL容量允差为±1.5%,100 μL容量允差为±2%。配置过程中使用1 000 μL移液器6次,使用200 μL移液器2次,100 μL移液器1次,按三角形分布计算标准不确定度,配置过程引入的相对标准不确定度为
=0.015 6
(3) 蛋白样品溶液浓度的相对标准不确定度为
=0.015 6
2.4.2 左、右摩尔吸光系数差值的不确定度
仪器经过校准,假定校准因子与波长无关,引入校准因子后的溶液吸光度值公式为:
ΔΑ0=xc0l0Δε0
(4)
ΔΑi=xciliΔεi
(5)
式(4)和式(5)中,校准因子x相等,因此可得蛋白样品溶液左、右摩尔吸光系数差值Δεi为
(6)
式(6)中,Δε0为校准样品d-10-樟脑磺酸铵的左、右摩尔吸光系数差值,是一个指定的文献值,没有相关不确定度[12]。
ΔA0和ΔAi分别为校准样品和蛋白样品的左、右摩尔吸光度,其标准不确定度是由仪器性能决定的,因此是一个统计值,在重复性的不确定度中已经进行评估。
c0和ci分别表示校准溶液的浓度和蛋白样品的浓度,溶液浓度的不确定度由样品的纯度及稀释过程引入,分别进行计算。ci的相对不确定度上文已经给出,同理计算c0的不确定度为
l0和li分别为校准溶液和蛋白溶液的比色皿光程长,其相对不确定度使用数据的相对标准偏差表示,li含有3个比色皿的数据,取其中最大值进行计算。
=0.057
2.5 0.05 mg·mL-1细胞色素C水溶液的圆二色值的合成标准不确定度
上述测定中各不确定度分量互不相关,则0.05 mg·mL-1细胞色素C水溶液的圆二色值的合成标准不确定度uθ(X)为

=0.270 mdeg
2.6 扩展不确定度U
取置信概率P=95%,包含因子kp=2,则其扩展不确定度为U=kp×uθ(X)=2×0.270=0.54 mdeg。
2.7 测定结果的表示
结果保留小数点后两位,0.05 mg·mL-1细胞色素C水溶液的圆二色值表示为:θ=(-4.53±0.54) mdeg,k=2,P=95%。将以上不确定度分量列于表3。
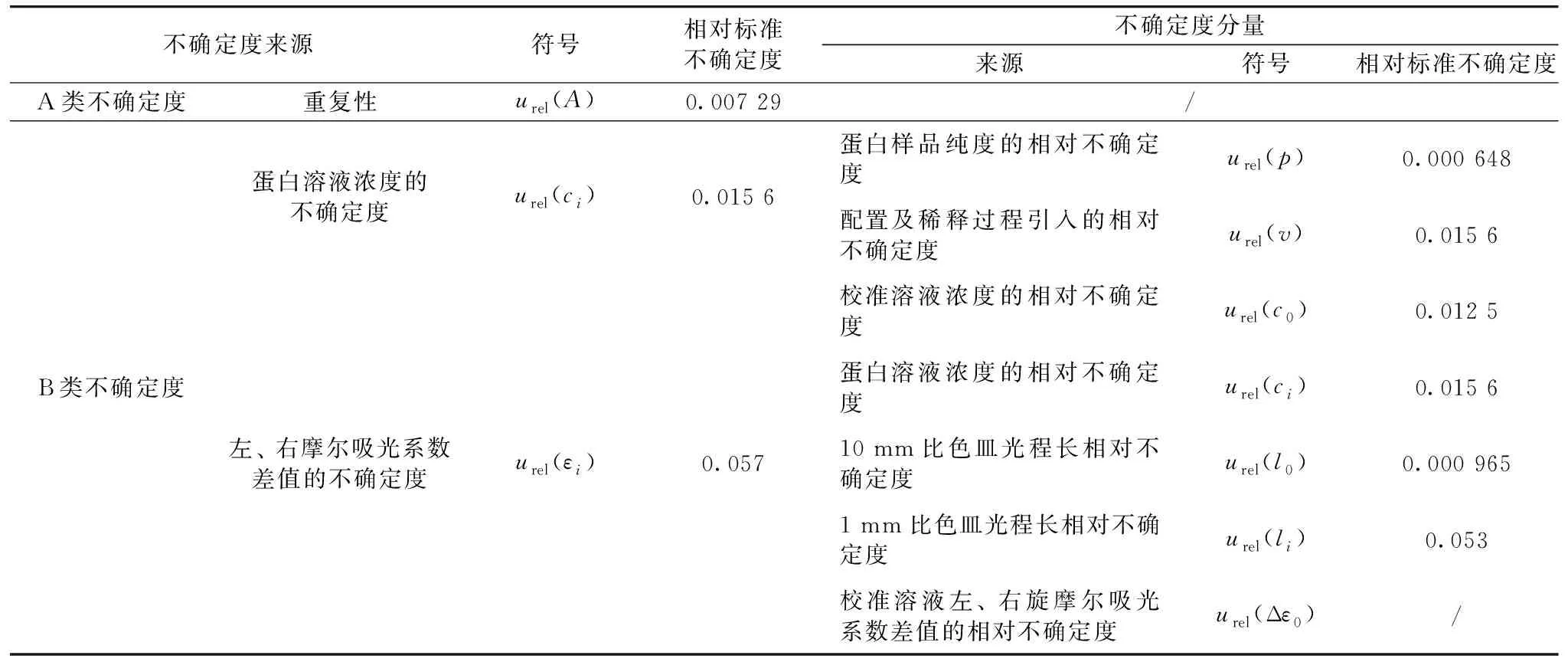
表3 不确定度分量列表Table 3 List of uncertainty components
通过表3可以看出,不确定分量最大的是1 mm比色皿光程长不确定度,在本文中该值使用测试数据的相对标准偏差进行计算,涉及谱图中谱峰强度和基线噪音,谱图的平滑等。真实的光程长可以用两种方法来测量[10,12],一是使用铬酸钾溶液在273 nm的吸收,二是干涉法,将比色皿放置在光谱仪中,并计算两个波长和之间的完整干涉条纹的数量。无论哪种方法测得,最终的光程长会受到样品粘度的影响。比色皿基线可能会受到残留的蛋白污染,如果谱图中基线偏移大于5 mdeg,则需要清洗比色皿后再行测试。30%盐酸+70%乙醇溶液,发烟硝酸或重铬酸钾-硫酸洗液均可以用来清洗比色皿中残留蛋白污染物[10]。选择高质量的比色皿也是获得高质量谱图的关键。
为了降低1 mm比色皿光程长不确定度,首先要使用高质量的或者有计量合格证书的比色皿;第二,在测试蛋白样品前,先测试空白溶液的CD谱,确认比色皿干净、基线偏移小于5 mdeg后再开始测试蛋白样品溶液;第三,选择合适的蛋白溶液浓度,不能过高也不宜过低。过高的浓度导致蛋白质分子间的聚集,但如果太稀,在样品溶液与玻璃移液管和试管接触中可能会因吸附在玻璃表面而导致蛋白质损失,影响测试的准确性。一般CD测量是在相对稀释的蛋白质溶液(0.01~0.2 mg·mL-1)上进行的,样品在感兴趣波长范围内的最大吸光度值不得超过2,最佳值为在信噪比最大的波长处吸光度值0.87;第四,保持CD光谱采集参数一致且合适,为了防止光谱变形,推荐经验的仪器参数设置选择为:扫描速度×响应时间<带宽(band width)<光谱特征宽度(一般蛋白质的宽度为15 nm)/ 10。比如本文采用的测试参数为:扫描速度50 nm·min-1,响应时间1 s,带宽1 nm。第五,保持处理CD光谱的参数一致。直接采集到无需处理的高质量CD光谱耗时较长,一般情况下,CD光谱需要经过平滑降噪处理后进行二级结构计算、解卷积分析或其他的比较。
另外一个较大的分量为溶液的不确定度,其中贡献占比大的是校准溶液和蛋白溶液的配置和稀释过程。在这个溶液配置过程中,尽量减少稀释次数,使用检定合格的容量瓶或移液器等可以减少其对不确定度的贡献。
3 结 论
CD光谱学是一种广泛使用的评估蛋白质的二级结构的技术,计算结果的可靠性与CD原始光谱图的质量直接相关。建立了CD光谱测试的简单模型,并使用它来评估一个α-螺旋主导蛋白质—细胞色素C在固定浓度0.05 mg·mL-1下的圆二色值的不确定度。这些不确定度既与随机因素有关,可以从重复测量中得出(A型评估),也与测试过程相关,只能采用非统计学方法进行评估(B型评估)。总体考虑了仪器性能、蛋白溶液浓度、校准溶液浓度、比色皿的光程长、左、右摩尔吸光系数带来的不确定度,并使用可靠的校正因子k=2,在对仪器进行校准的情况下,评估了0.05 mg·mL-1细胞色素C在波长为222 nm处的圆二色值不确定度,为±0.54 mdeg。通过不确定度的评定,找出影响测量结果准确性的主要因素,其中较为显著的不确定度分量为1 mm比色皿光程长不确定度和溶液配置、稀释过程引入的不确定度,设法消除或降低这些因素的影响,可以改进测量方法。以测量不确定度的评定为工具,解剖分析测量过程,探讨CD数据的可比性和可靠性,为准确评估蛋白质的二级结构奠定基础,并为进一步开展CD光谱的实验室间比对提供实验参考。
猜你喜欢
娃娃乐园·综合智能(2022年9期)2022-08-16
科学大众(2021年9期)2021-07-16
山东化工(2019年24期)2020-01-17
天文研究与技术(2019年4期)2019-10-23
实验技术与管理(2019年3期)2019-04-03
商品与质量(2018年47期)2018-12-07
科学家(2017年8期)2017-06-22
中国调味品(2017年2期)2017-03-20
现代食品(2016年24期)2016-04-28
应用光学(2015年3期)2015-06-10
