
过渡金属催化氯代物的羰基化反应研究进展
2023-10-14 07:52王鹏史会兵赵德明冯保林陈倩杨妲
化工进展 2023年9期
王鹏,史会兵,赵德明,冯保林,陈倩,杨妲
(1 山东京博石油化工有限公司, 山东 滨州 256500;2 中国石油大学(华东)理学院,山东 青岛 266580)
羰基化反应能够在底物分子内高效构建羰基官能团而被广泛关注,通过简单的底物(烯烃、炔烃、烷烃、卤代烃等基础化学品)可制备多种高附加值化学品[1-9]。其中有机卤代物作为底物也可制备结构多样性的大宗化学品、精细化学品以及关键医药中间体[10-11]。有机卤化物发生羰基化反应的活性顺序不同,具体为C—F≪C—Cl<C—Br<C—I活性依次增加(碳卤键能影响),可以看出C—Cl键的活性相对较低而使其在温和条件下难以高效高选择性转化[12],但是氯代物相对于溴代物和碘代物价格低廉,因此如何研发或者改善现有催化剂体系,实现氯代物温和条件下的高效转化或者拓宽更多类型氯代物的羰基化反应成为科研热点[13-14]。本文综述近年来不同类型氯代物与一氧化碳(CO)、各种亲核试剂发生羰基化反应合成高附加值化学品的研究进展,同时对氯代物羰基化反应依然存在的挑战和未来应用前景进行了展望(图1)。
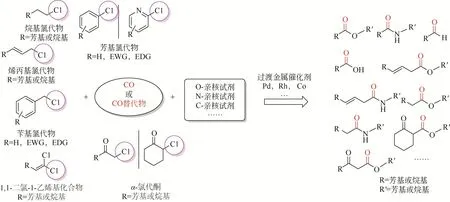
图1 有机氯代物的羰基化反应转化
1 氯代物羰基化反应机理
在CO 作为羰源的条件下,芳基卤化物的氧化加成机理不同于钯催化偶联过程中相同芳基卤代物的氧化加成机理[1,15-16]。这种差异是由无CO 配位的活性Pd(0)络合物和有CO 参与配位的非活性Pd(0)羰基加合物(或簇)之间的含量不平衡造成的[1]。虽然双齿膦配体可以有效阻止Pd(0)化合物的聚集,但在CO 存在的条件下,膦配体以及CO 分子(一个或两个)依然可以和Pd(0)配位形成包含桥接CO 部分的双金属二聚体,从而影响反应活性[1,17]。
以钯催化剂为例,氯代物发生羰基化反应的机理如图2。首先是CO、配体氛围中钯络合物的形成,其次经历氯代物与钯催化剂的氧化加成,再次CO 配位及插入形成酰基钯活性中间体,在这一步,CO 迁移插入方形平面芳基氯化钯配合物的速度很快,并且已被提出通过五配位中间体发生[1,18]。最后发生亲核试剂进攻,钯催化剂还原消除,同时反应体系中的碱缔合氯化氢而形成目标产物。

图2 钯催化芳基氯代物与亲核试剂羰基化反应机理
在CO存在下,氯代物与Pd(0)氧化加成的机理研究报道很少。但是由于氯化物相比其他卤化物便宜并且更具商业性,因此其羰基化反应也是研究热点[19-20]。研究发现,芳基氯化物参与Pd(0)氧化加成的步骤以及羰基化反应本身的速率都很慢,这是因为芳基氯化物与低配位、富含电子的Pd(0)物种发生氧化加成反应速率最快,而CO 的结合使电子云密度较低,配位数增高,因此也抑制了反应速率[1,20-22]。由于芳基或杂芳基氯化物的羰基化需要较高温度(>100℃),这也可能会导致CO 从Pd(0)配合物中解离或Pd(0)羰基簇的解离[1,23-26]。因此,更好地了解与芳基氯代物反应的金属配合物的特性,可以发现新的催化剂体系,从而诱导该类底物的温和羰基化转化。
2 芳基氯代物的羰基化反应
芳基氯代物的羰基化转化由于其价格廉价且产物用途广泛获得而备受关注[27-37],然而C(sp2)—Cl键较高的离解能导致其相对于溴代物和碘代物更难活化(室温条件下,PhCl、PhBr和PhI的C(sp2)—X键能分别为402kJ/mol、339kJ/mol和272kJ/mol)[28-32]。
2.1 甲酰化反应
早在1982 年,Pétrier 等[33]报道了温和条件下N,N-二甲基甲酰胺(DMF)作为羰源的芳基氯(氯苯)和烷基氯(1-氯丁烷、环己基氯)的甲酰化反应合成醛类化合物。经过条件筛选,当四氢呋喃(THF)作为溶剂时,目标产物的收率为70%~78%。2021年,Xiao等[34]也报道了一例DMF作为羰源(同时作为溶剂),CuI 催化氯苯可实现甲酰化高效合成苯甲醛(收率86%),同时该催化剂体系对烷基/芳基溴代物具有中等到优异的甲酰化底物普适性(45%~96%)。
2015 年,Iranpoor 等[35]首次使用五羰基铁作为羰源实现了钯催化芳基卤化物的甲酰化反应合成芳香醛和氘代醛。该反应在常压条件下以DMF 的水溶液作为溶剂,不需要添加气态氢或任何还原剂便可生成醛类化合物。有趣的是芳基碘代物可在无膦配体的情况下发生反应[式(1)]。
另一方面,也有CO 作为羰源的研究报道,例如1989 年,Huser 等[36]报道了钯催化芳基氯代物的甲酰化反应。当氯苯作为底物时,经过条件筛选(trans-[Pd(PC6H11)3)2(C6H5)Cl]/三环己基膦作为催化剂,添加三乙胺(Et3N)作为碱,甲苯作为溶剂,1.5MPa 合成气),苯甲醛的收率高达90%。同年,Ben-David 等[37]也报道了一类富电子并且螯合稳定的钯催化剂(dippp)2Pd(0)催化氯苯的甲酰化反应,该体系选择甲酸钠替代氢气可实现苯甲醛95%的高效合成。
2.2 羰化胺化反应
在已知制备芳香酰胺的各种方法中,芳基碘化物和溴化物与CO、N-亲核试剂的羰化胺化反应报道较多[38-45]。而使用芳基氯化物作为起始原料的报道相对较少[27,46-47],该领域的大部分工作均使用缺电子(或N-杂)氯代芳烃作为起始原料[40,48-54]。目前为止,只有两种方案(一步法策略和串联策略)应用于氯苯或含有给电子基团的芳基氯化物的羰化胺化反应[15,55-58]。
对于一步法策略,文献报道中大多选用氨气(NH3)作为亲核试剂,而其他类型的伯、仲脂肪族和芳香族胺作为亲核试剂的报道极少[15,55]。2010年,Wu 等[55]报道了钯催化芳基和杂芳基卤化物(22 个实例)与CO、氨气(NH3)作为胺源的羰化胺化反应。该催化剂体系同样适用于非活化的芳基氯化物(8 个实例),在温和条件下以30%~60%的收率生成所需目标酰胺[条件1,式(2)]。2018 年,Wang 等[15]探究了乙烯基钯络合物(DCPP)Pd(C2H4)作为催化剂催化芳基氯化物与氨气的羰化胺化反应的机理。该工作中,氯苯以83%的产率转化为苯甲酰胺,缺电子/富电子的芳基氯化物均可以64%~100%的产率生成相应的苯甲酰胺[条件2,式(2)]。
对于串联策略,2007年,Martinelli等[56]开发了一种通用的、耐官能团的、温和的体系,用于钯催化芳基氯化物串联合成酰胺的羰基化反应。该反应在低CO压力(0.1MPa)和中等温度(110℃)下进行,通过酚钠的引入,一方面促进了苯甲酸酯作为反应中间体的形成,另一方面可以作为随后与胺反应的离去基团[式(3)]。该体系所选用的膦配体(DCCP·2HBF4)廉价、在空气中稳定且商业可得,具有潜在的工业化放大前景;另一方面,他们通过原位红外光谱详细探究了酚钠在反应过程中的作用。
2018 年,Lagueux-Tremblay 等[57]利用串联的策略报道了钯催化芳基氯化物和4-二甲氨基吡啶(DMAP)的羰化胺化反应。该反应首先生成芳酰基DMAP 盐作为强亲电体,其次伯胺和仲胺的亲核进攻生成具有良好官能团相容性的酰胺[式(4)]。与通常需要亲核试剂与弱亲电钯酰基中间体反应的经典羰基化反应不同,芳酰基DMAP 盐的高亲电性允许多种底物的酰化。这种转化是由“Pd/Xantphos”催化剂介导的,机理研究表明,膦配体位的空间位阻效应影响其与Pd(0)的结合,从而影响ArCO-DMAP 产物的还原消除和高键能芳基碳氯键的氧化加成过程。总之,这种转化允许芳基氯化物与一系列亲核试剂以良好的官能团相容性生成酰胺和酯。
2022 年,Wang 等[59]首次报道了钯催化低活性芳基氯化物(包括富电子、中性和缺电子的芳基氯代物)与伯、仲脂肪族和芳香族胺的直接羰化胺化反应。 该转化成功的关键是在“Pd(OAc)2/Xantphos”催化剂体系中引入氯化铯(CsCl)作为添加剂。该催化体系实现了多种芳基氯化物与胺的有效转化合成酰胺,并具有良好的官能团相容性(60 个实例,转化率高达99%,分离收率高达95%)[式(5)],同时也有效改善了该类型的底物需要通过串联策略合成目标产物的弊端。研究表明,亲核试剂胺的用量会影响一氧化碳迁移形成酰基钯中间体的过程,过量的胺会抑制羰基化产物的生成;并且该反应体系中一氧化碳的压力(0.2MPa)过高或者过低均不利于反应的发生。
2.3 羰化酯化反应
醇通常比胺酸性更强,因此在碱性条件下,过渡金属催化芳基氯代物可以原位生成醇盐,并且醇或醇盐可作为偶联试剂继续发生后续反应。相反,胺作为亲核试剂很难形成阴离子酰胺盐。另一方面,醇和醇盐的配位能力和亲核性与胺的也不相同,这便导致酰基配合物与氧基亲核试剂的反应机理与氮基亲核试剂的反应机理有所不同,发生羰基化反应的难易程度也有所差异[15,58]。
早在1989 年,Ben-David 等[58]报道了钯催化芳基氯代物的羰化酯化反应。该工作选择 (dippp)2Pd作 为 催 化 剂 [dippp=bis(diisopropylphosphino)propane],可以高效高选择性合成芳基羧酸、酯和酰胺类化合物。随后1990 年,Dufaud 等[60]也报道了钯催化芳基氯代物(氯苯和缺电子取代的氯苯)的羰化酯化反应制备多种酯类化合物,Pd/C 作为催化剂,但是反应条件苛刻,200℃反应50h 才能实现底物的转化,因此发展较温和条件下芳基氯代物的羰化酯化反应显得尤为重要。
2001 年,Mágerlein 等[52]报道了氯苯类化合物的高效羰化酯化反应。该工作发现了一种新的钯催化剂体系以良好到优异的收率催化缺电子、中性和富电子的芳基氯化物生成目标产物。所使用的配体商业可得,并且在空气中稳定(条件1,图3)。次年,Mägerlein 等[61]优化反应条件,发现“PdCl2(PhCN)2-PCy3”组成的催化剂体系亦能很好的催化芳基氯代物的羰化酯化反应(条件2,图3)。所选用的膦配体“三环己基膦”更加廉价易得,且目标产物收率优异(68%~91%)。

图3 芳基氯化物羰化酯化反应的高效催化剂
2010 年,Schareina 等[62]选择一氧化碳替代物,实现了钯催化芳基氯代物的羰化酯化反应。经过条件筛选:1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)作为碱,乙腈作为溶剂时,目标产物酯收率高达81%。在对CO 替代物筛选的过程中发现:甲酸乙酯作为羰源时,3,4-二甲基苯甲酸丁酯收率为56%,而甲酸异丙酯作为羰源时,由于空间位阻效应目标产物收率较低(34%)[式(6)]。另一方面,该策略将底物普适性拓展至含有富电子基团的芳基氯代物。
2017 年,Shimomaki 等[63]以CO2作为羰源,在可见光的驱动下,实现了钯催化芳基卤化物的羰化酯化反应。该催化剂体系可以避免使用当量的金属还原剂,且底物普适性优异;对于芳基溴代物亦可以高效转化,首次实现了大位阻的2,4,6-三异丙基溴苯的羰化酯化反应[式(7)]。
2020年,Ai等[64]报道了一种新型钯催化芳基氯化物的羰化酯化方法以良好至优异的产率合成多种苯甲酸甲酯。该反应能够发生主要有以下因素:
①使用LiOMe可以作为促进羰基化转化的碱或共亲核试剂;②使用Pd/C 作为催化剂,可以防止传统的二价钯前体被甲醇还原为零价钯而团聚;③CO浓度不能过高,否则将直接导致反应终止[式(8)]。该策略的优势在于通过一种催化剂体系实现了各种类型的芳基氯代物(包括富电子、中性和缺电子)羰化酯化反应一步法合成酯。
3 烯丙基氯代物的羰基化反应
烯丙基氯代物与CO 羰基化反应转化为β,γ-不饱和酯或酰胺的报道较少[65-73]。早在1963年,Tsuji等[66]报道了化学计量的π-烯丙基氯复合物可以调控烯丙基氯在乙醇溶剂中与CO 结合发生羰基化反应生成丁烯酸酯。很快他们[67-73]发现催化量的氯化钯也可以使烯丙基氯、烯丙基醇、烯丙基醚等作为底物促进该反应[式(9)]。
1964 年,Dent 等[74]报道了π-烯丙基钯配合物催化烯丙基氯化物的羰基化反应。该反应收率优异(94%)[式(10)]。随后1969年,Medema等[75]报道了这个反应的详细动力学研究。但是该反应需要在较高温度(130℃)下发生,同时催化剂用量较大,很难实现工业化应用。
1995 年, Yamamoto[76]报道了甲酸钾参与钯催化肉桂酰氯的羰基化反应合成β,γ-不饱和羧酸。反应通过18-冠-6活化甲酸钾(HCOOK),反应温度50℃较温和,但是CO压力5.0MPa偏高[图4(a)]。同时,在低温条件下使用PdCl2(PPh3)2可以实现2-取代烯丙基氯化物的双羰基化过程[室温条件二乙胺作为亲核试剂,图4(b)]。
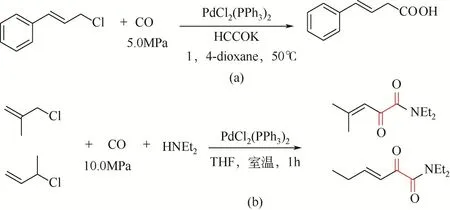
图4 钯配合物催化烯丙基氯化物的羰基化和双羰基化反应
2014 年,Zhang 等[77]首次报道了铱催化烯丙基氯化物的羰化胺化反应高效高区域选择性合成氨基甲酸酯(支链产物与直链产物的摩尔比高达98/2)。该反应羰源为二氧化碳(0.1MPa)且反应条件温和(三乙烯二胺DABCO 作为碱,甲苯作为溶剂,反应温度15℃)[式(11)],但是产物的区域选择性调控依然是难点。
2017 年,Wu 等[78]报道了温和条件下,甲酸作为羰源,钯催化烯丙基氯羰基化直接合成β,γ-不饱和羧酸的反应(60℃)。虽然这类底物比其他烯丙基衍生物具有更好的产率和线性选择性,但底物范围受到限制[式(12)]。
2022 年,Wang 等[79]报道了钯催化烯丙基氯代物为底物的羰基化反应(羰酯化和羰化胺化)制备β,γ-不饱和酯/酰胺。该工作通过对关键反应参数(钯催化剂前体、溶剂和碱等)进行筛选优化,可以在温和条件下[Pd(OAc)2摩尔分数0.1%、CO 压力0.2MPa、60℃]合成多种β,γ-不饱和酯/酰胺。该催化剂体系对烯丙基C—Cl键的活化表现出优异的化学和区域选择性(图5),产物中没有支链的羰基化产物生产,且无需膦配体参与、催化剂用量较低,有望实现工业化应用;另一方面,该策略对于不同类型的亲核试剂(醇、酚、脂肪胺、芳香胺)具有良好的兼容性,亦能实现复杂天然产物分子的改造合成。
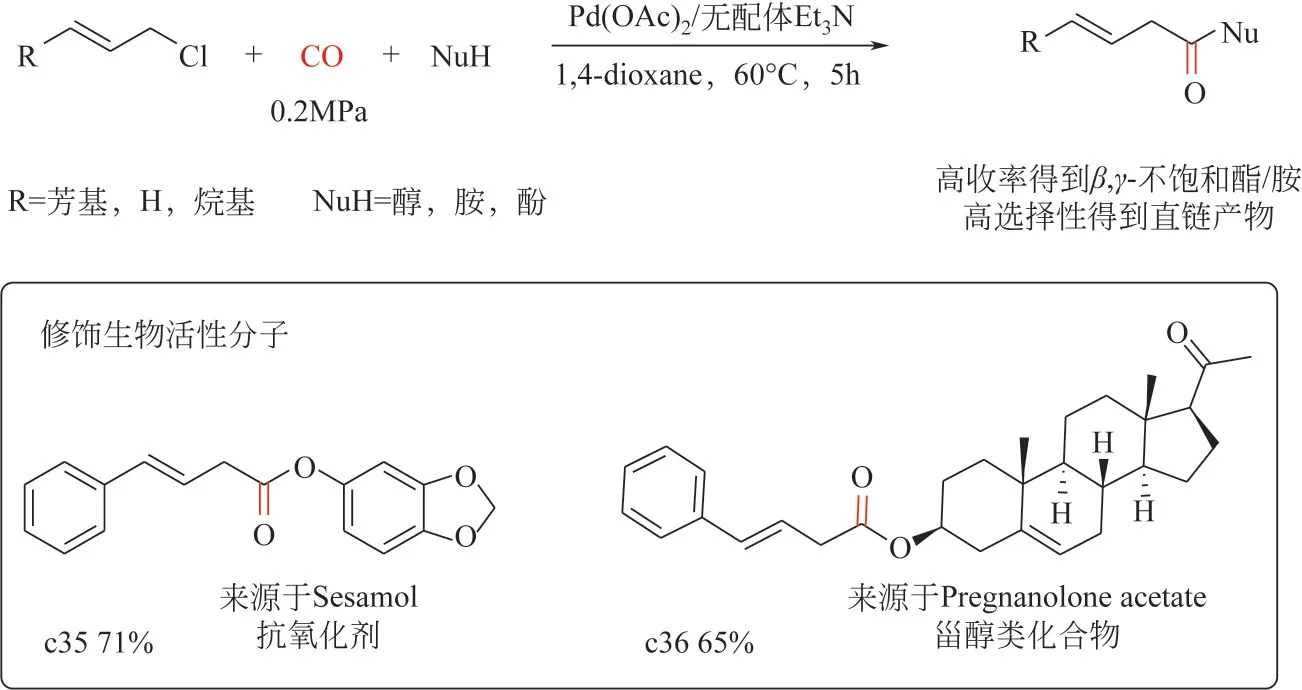
图5 钯催化烯丙基氯羰基化制备β,γ-不饱和酯/酰胺
4 苄基氯代物的羰基化反应
4.1 苄基氯代物羰基化反应
早在1974年,Schoenberg 等[80]报道了一类“乙酸钯-三苯基膦”配合物在叔胺为碱的条件下催化氯化苄与CO、丁醇的羰化酯化反应。随后,Hidai等[81]报道了钯催化溴化苄的羰化酯化反应,钯催化剂为Pd(CO)(PPh3)3、Pd3(CO)3(PPh3)4、Pd3(CO)3(PPh3)3和PdCl2(PPh3)2等。虽然该反应条件中需要引入一种以上的当量碱中和卤化苄反应过程中产生的HX(X为卤原子)。当然也有无碱反应体系的报道,例如,Urata 等[82]报道了分子筛类的沸石可以作为卤化氢的有效吸收载体。
1983 年,Foà 等[83]报道了钴催化苄基氯的羰化酯化反应,该催化剂体系65℃反应6h 收率高达65%。随后1984 年,Tustin 等[84]报道了铁催化苄基氯代物的羰化酯化反应。当甲醇作为亲核试剂,碳酸钾做为碱时,目标产物的收率高达65%。该反应在常压室温条件下反应过夜即可发生[式(13)]。
1989 年,Adapa 等[85]报道了温和条件下苄基氯代物羰基化反应生成苯乙酸叔丁酯。作者对1-溴乙苯的羰基化反应也进行了研究[86],与氯化苄不同的是溴化苄对酯的选择性较低。由于该类底物为仲碳取代的卤代物,副产物除醚类化合物外,还可以在碱性条件下发生消除反应生成一定量的苯乙烯(10%~25%)。反应通过添加过量的配体,酯的收率可提高至70%[式(14)]。
除了合成酸和酯类化合物,卤化苄可与其他亲核试剂发生羰基化反应合成酮、酰胺等化合物。在磷酸钾存在的情况下,Wu 等[87]利用商业可得的Pd(OAc)2/PCy3催化剂体系,报道了苄基氯化物与芳基硼酸的羰化偶联反应生成α-芳基苯乙酮。值得注意的是,该反应以水作为溶剂[式(15)]。随后,Wu 等[88]将亲核试剂扩展到三氟芳基硼酸钾,可以避免偶联副产物的生成。
2011 年,Wu 等[89]成功地将苄基氯化物应用于钯催化末端炔烃的羰化反应中。该反应中芳香族炔烃与脂肪族炔烃都具有很好的普适性。当苄基乙炔作为偶联剂时,1,4-二芳基-3-丁-2-酮或取代呋喃酮的产量较好。该反应成功的关键是使用了缺电子的亚磷酸酯配体(图6)。

图6 苄基氯化物与末端炔烃的羰基化偶联反应
2010 年,Troisi 等[90]发现在合成β-内酰胺的反应中目标产物收率较低,有大量相应的非环状酰胺生成。他们认为是亚胺分解为胺类化合物导致的,胺进一步与酰基钯中间体反应生成酰胺。基于这一发现,他们开发了一种钯催化由苄基卤代物和胺羰基化合成酰胺的新方法。反应形成酰基卤化钯中间体后,经历来自胺的酰基亲核取代反应而生成目标产物酰胺。当苄基氯代物作为底物时,酰胺的选择性高达97%;烯丙基氯代物作为底物时,目标产物选择性高达92%[式(16)]。
2012年,Wu等[91]报道了氨气作为胺源的Pd催化氯化苄的羰化胺化反应。该反应通过对膦配体和催化剂前体的筛选,以中等至优异的收率(75%~98%)合成了不同类型的一级酰胺类化合物。在这个反应中,氨具有双功能性:不仅用作胺化试剂,而且用作碱[式(17)]。
2020 年,Richardson 等[92]报道了钯催化苄基氯代物的羰化胺化反应。该反应通过对溶剂、碱以及催化剂前体进行筛选,使用廉价的膦配体(DPEPhos)在较低温度(70℃)下实现了底物与伯胺/仲胺的羰基化反应,该反应能够抑制偶联副反应的发生,高效高选择性地合成2-芳基乙酰胺类化合物(图7)。该催化剂体系中膦配体亦商业可得,且底物普适性广泛,具有较好的官能团兼容性,为复杂的化学品的合成提供了新的方法。

图7 钯催化苄基氯代物的高效羰化胺化反应
在合成杂环化合物方面,基于卤代烯丙基底物和亚胺的[2+2]环加成羰基化工作[93]:2009 年,Troisi 等[94]报道了钯催化卤化苄化合物羰基化和亚胺立体选择性合成3,4-二芳基β-内酰胺;苄基溴的反应速度比苄基氯化物快,但β-内酰胺产率接近[图8(a)];2013年,Wu等[95]报道了以水杨酸醛和苄基氯化物为原料,钯催化羰基合成色甲酮的方法,以中等到优异的收率分离出多种香豆素(30%~95%)[图8(b)]。
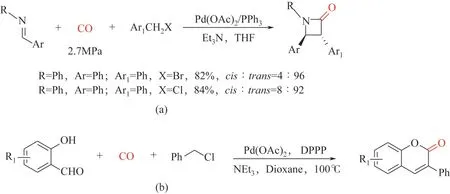
图8 以卤化苄为底物合成杂环化合物3,4-二芳基β-内酰胺以及色甲酮
4.2 氯化苄羰基化反应的工业化应用
苄基氯代物羰基化反应关注度最高的底物为氯化苄,其下游的苯乙酸以及苯乙酸酯类化合物在生活生产中具有重要的用途,例如可以制备重要医药青霉素工业盐(PGK)如阿莫西林、头孢氨苄、头孢拉定、舒巴坦、他唑巴坦等抗生素的中间体[96-103]。工业生产中,苯乙酸的生产方法主要有苯乙腈水解法、苯-醋酐法、乙苯氧化法、苯-甲醛羰基化法、苯乙酰胺水解法等多种策略[96,98-100]。但是都存在以下弊端:反应过程中副产物较多,对操作环境污染严重,产品收率低,生产成本高。而苄氯羰基合成法(苄氯在氢氧化钠和有机溶剂两相体系中进行羰基化反应,生成苯乙酸钠,然后在酸性条件下被酸化成苯乙酸)可高效制备苯乙酸而备受关注。该策略相较于传统通过氯化苄和氰化物反应得到目标产物的方法具有如下的优点:①避免使用氰化钠及中间体苯乙腈等剧毒物品,同时可以避免苯乙腈合成工序中产生易挥发剧毒且带有恶臭的物质异氰苄,减少人员健康危害,减少这些物质对环境造成严重污染;②有效改善反应过程中的废水污染;③改善苯乙腈合成过程中收率低的弊端[97,99-103]。但是该反应以钯催化剂为主,由于催化剂价格昂贵,如何解决羰基化过程中面临的成本问题便成为研究重点[98-101]。最主要的思路便是利用两相体系实现原料、催化剂与产物分离,进而实现催化剂的多次循环利用。
Cassar 等[96]报道了钯催化两相体系中卤化苄发生羰基化反应一步法生成相应的酸。该策略将氯化苄和PPh3缓慢加入氢氧化钠(30%)的水溶液中,同时加入Pd(PPh3)2Cl2和n-Bu4I,在95℃、CO 压力0.5MPa 条件下制备苯乙酸,产率高达83%。值得注意的是,该反应体系的催化剂可以多次循环使用而没有明显的流失(图9),因此该策略有望实现原料、催化剂与产物的分离,并且大规模生产应用。
Okano 等[97]在Cassar 两相催化剂体系的基础上报道NaOH/庚烷两相体系,使用磺化的膦配体将钯络合物溶解在水相中。该方法后续被Kohlpaintner等[98]进一步发展,使用Pd(OA)2(或PdCl2)以及磺化膦配体,催化氯化苄在温和条件(CO 压力0.1MPa)下产生相应的苯乙酸,产率为80%~94%。反应后水相经有机溶剂萃取便得到羧酸钠盐[转化数(TON)>1500;转化频率(TOF)达到135h-1]。随后经过酸化产生游离酸产物(图10)。该策略很好地实现原料、催化剂与产物分离。
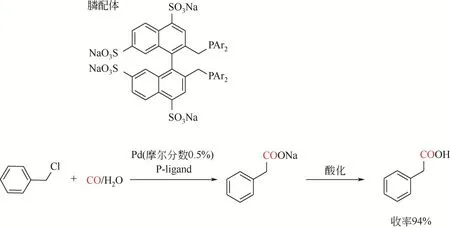
图10 钯催化苄基氯代物羰化酯化反应合成苯乙酸
1999 年,Pellegrini 等[99]除了使用CO 和甲 醇使氯化苄进行羰基化反应外,选择CO 替代物甲酸甲酯作为羰源,Mg(OMe)2诱导甲酸甲酯脱碳促进反应的完成。他们发现添加额外的CO 会抑制羰基化过程[图11(a)]。随后2001年,Gaviño等[100]通过优化反应条件再次报道了温和条件下钯催化苄基氯化物的羰化酯化反应,并且探究了CO 压力对该反应的影响。他们在没有CO 的情况下加热反应介质(避免形成钯羰基物种,高浓度的该物种不利于羰基化过程),利用镁形成甲氧基离子,再通入CO,便可以在较低的CO 压力(30℃、0.3MPa)和适当的搅拌速度下,避免催化剂失活从而促进反应的发生[图11(b)]。2006年,Pryjomska 等[101]也报道了一例较温和条件下(60℃、CO 压力为0.5MPa)钯催化苄基氯的羰化酯化反应,但是产物苯甲酸甲酯的收率较低(15%)[图11(c)]。如何在温和条件下提高目标产物的收率也是该策略的研究难点。

图11 温和条件下苄基氯的羰化酯化反应
除了钯催化剂体系,钴系催化剂亦能很好地催化苄氯羰基合成苯乙酸[102-106],该催化剂体系已经实现工业化应用。当Co2(CO)8作为催化剂前体,反应可在较温和(30~70℃)、CO 压力0.1~1.0MPa 条件下发生,苯乙酸收率可达85%~90%。当四羰基钴的钠盐为催化剂前体时,反应在水油两相中进行,生成苯乙酸钠中间产物,经过酸化后得目标产物,收率一般为80%~90%。选择适当的助剂可使苯乙酸钠不断地从水相中分离出来,催化剂完全溶于有机相中循环,从而实现生产过程的连续化。该方法的优点如下:①催化剂制备贮存及循环较容易,回收率高;②羰基化反应在低压条件下即可发生;③苯乙酸收率较高,重结晶后产品纯度高;④CO 利用率高,三废较易处理(其中废盐水经处理可用于氯碱厂作电解原料)[103]。
总之,为解决过渡金属催化剂分离和循环使用的方法(均相催化多相化)可归纳为两大类:一类是将催化剂静态固定在高分子或无机载体上,形成固载化均相催化;另一类则是采用功能化的膦配体(水溶性膦配体或离子化的膦配体),将均相催化剂动态锁定在与产物互不相溶的水相(或者离子液体)中,而实现水/有机两相催化[107-108]。而该方法也为从源头上解决金属催化剂回收及三废后处理等问题提供一个有效的工具[108]。另一方面,陈金龙等[109]也报道了一种苯乙酸生产废水的治理与资源回收利用方法,它是将苯乙酸以及生产后废水通过苯乙烯-二乙烯基苯共聚大孔吸附树脂,使废水中苯乙酸等大部分有机物吸附在树脂上;吸附出的水无色透明,可送隔膜法电解车间配制电解盐水以回收利用其中的氯化钠。
5 α-氯代酮的羰基化反应
[Pd]/P配体体系催化的α-溴代丙酮的羰基化反应(特别是羰化酯化)在1975 年就有报道[85,110], 但是α-氯代丙酮的羰基化反应报道较少[13,111-113]。1989年,Adapa 等[85]报道了温和条件下芳基卤代甲基衍生物的羰基化反应制备叔丁酯,该工作只有一个实例为α-氯代丙酮羰基化生成相应的酯(收率为50%)。1999 年,Cavinato 等[114]报道了NEt3作为碱,PdCl2(PPh3)2-PPh3催化α-氯代环己基酮羰基化反应合成β-酮酯类化合物。该催化剂体系需要较高的CO压力(10.0MPa),并且收率伴随CO压力的增加而增多[式(18)]。
2002 年,Lapidus 等[115]报道了三丁胺存在下,钯催化卤代甲基酮(X=Cl,Br)的羰基化反应合成乙酰乙酸烷基酯和β-芳基-β-酮酯类化合物,产率为68%~86%。该反应条件为CO 压力1.0MPa,110℃反应2h完成[式(19)]。该反应体系对于氯甲基酮具有良好的羰基化化学选择性,当2-溴苯乙酮做底物时,可产生大量的苯乙酮作为副产物。通过延长反应时间,该反应可以在一个大气压条件下发生。他们对反应机理进行了探究:羰基化过程中存在碱(Bu3N)不仅可以中和形成的氢卤酸,而且可以催化酰基络合物的醇解;另一方面,Bu3N 的浓度对反应速率的影响表明酰基络合物的醇解是速率限制步骤。
2012 年,Wahl 等[116]采 用Pd(acac)2/Xantphos 催化α-氯酮发生羰基化反应合成β-酮酯,目标产物收率高达98%,该催化剂体系底物普适性良好,适用于不同类型的醇类、一级和二级α-氯酮类底物[式(20)]。他们通过对反应条件的优化,以使用比文献报道的低得多的催化剂负载量(摩尔分数0.1%)高产率生成β-酮基酯,通过高压核磁、红外表征以及理论计算探究了催化循环过程中的活性中间体,并验证了膦配体(Xantphos)的选用有助于提高目标产物的收率,是相较于碱、一氧化碳压力、温度等因素中最重要的影响反应活性的因素。
2014 年,Perrone 等[117]报道了芳香族亚胺和α-氯酮的立体选择性羰基化反应合成具有N-芳基或N-烷基取代的α-亚烷基β-氧代酰胺。该反应可以一锅法高立体选择性生成Z式异构体的目标产物酰胺(24%~80%)[式(21)]。该方法可应用于含有N-芳基或N-烷基取代基的多种C-芳基亚胺,整个过程是高度立体选择性的,并且仅提供作为(Z)异构体的α-亚烷基β-氧代酰胺,还提出了一种涉及酰基-β-内酰胺中间体的机制假说。
羰化串联反应能够以氢甲酰化的产物醛为中间产物进一步合成高附加值化学品,与多步反应相比,一锅法串联反应更为绿色和经济。α-氯酮的羰化串联反应由于能够合成更高附加值产品也引起了广泛关注[118-120]。2012 年,Wahl 等[121]报道了钯催化α-氯酮串联羰基化/烯丙基化加成反应合成烯丙基酮酯类产物。在第一步中,α-氯酮羰基化形成β-酮酯作为亲核中间体,然后在4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(Xantphos)配体的调控下,与烯丙基苯甲酸酯反应以良好的产率获得目标烯丙基化β-酮酯[图12(a)]。同年,Giboulot 等[122]报道了钯催化α-氯酮串联羰基化/脱羧烯丙基化反应高选择性合成γ,δ-不饱和芳香酮。该反应中,α-氯代苯乙酮首先与烯丙醇发生羰基化反应,然后中间产物原位脱羧生成相应的单烯丙基酮。该反应底物普适性优异,底物范围可扩大到取代的α-氯代苯乙酮以及各种烯丙醇[图12(b)]。

图12 钯催化的α-氯酮参与的羰化串联反应
2013年,Wahl 等[123]报道了一类以α-氯酮为底物,通过简单高效的串联羰基化/迈克尔加成反应制备α-烷基β-酮酯类产物(图13)。他们通过条件优化可以在一锅反应中使用相同的碱以良好的产率(高达86%)制备高度官能化的α-烷基化β-酮酯,包括12 种原始产物。该多米诺反应的范围可以扩展到具有各种迈克尔受体的伯和仲氯酮,包括丙烯酸甲酯、甲基乙烯基酮和环烯酮等。

图13 α-氯酮的羰基化/迈克尔加成串联反应
6 1,1-二氯-1-乙烯基化合物的羰基化反应
文献报道在碱存在的情况下,烷基四羰基钴配合物RCo(CO)4可在温和条件下催化芳基卤化物的羰基化反应,从而高效合成烷基芳基酮、羧酸和α-氧代羧酸的混合物。该反应也被应用于卤代乙烯的羰基化反应,得到相应的羧酸。早在1989年,Miura等[124]报道了氢氧化钠作为碱,八羰基二钴与碘甲烷反应原位生成的四羰基甲基钴作为催化剂,催化1,1-二氯-1-乙烯基化合物进行羰基化反应,高效合成丙烯酸类化合物。当甲醇和水作为溶剂时,目标产物选择性高达68%。
后来,α-卤代丙烯酸酯被广泛应用于有机合成和医药中间体的生产中,后续可进行多种转化,例如发生迈克尔反应;环加成合成噁唑、吡唑或环谷氨酸盐;α-卤代丙烯酸酯的C—C双键也可以对映选择性氢化产生α-氯代、α,β-不饱和醛或烯丙醇[125-129],这些通过转化得到的高附加值产品已用于制备药物或用于天然产物合成中[130-133]。但是高效合成α-氯代丙烯酸酯的报道很少:Wittig 反应、Horner Wadsworth-Emmons 反应、氯甲硅烷基烯酮衍生物与醛反应制备等[134-137],通过羰基化反应策略制备的案例更少。
2010年,Arthuis等[138]以1,1-二氯-1-烯烃为底物,通过钯催化的羰化酯化反应高效合成了一系列Z-α-氯代丙烯酸酯。反应条件温和(CO 压力常压、70℃、24h),目标产物收率55%~91%[式(22)],该反应具有良好的官能团兼容性以及广泛的底物普适性。但是对于1,1-二氯-1-烯烃为底物的羰化胺化反应、甲酰化反应均没有报道。
7 烷基氯的羰基化反应
相比于芳基氯代物的羰基化反应,烷基氯代物的羰基化反应报道很少。经过文献调研,烷基碘化物和溴化物的羰基化反应已有大量报道,而过渡金属催化的烷基氯化物作为底物的羰基化反应仍然是此类转化的挑战[4,139]。
对于烷基氯代物的羰基化反应,早在1989年,Huser 等[140]就报道了二氯甲烷参与钯催化羰基化反应的案例,同时也提到了羰基化反应过程中对氯苯的活化。但是烷基氯代物的氢甲酰化反应还没有报道,目前只有烷基溴代物和碘代物的氢甲酰化反应报道[141-142]。2007年,Jia等[143]报道了KI存在下,钴配合物[Co(OAc)2、CoCl2]催化氯代烷烃与CO的羰化酯化反应。结果表明,Co(OAc)2的催化活性高于CoCl2,碱性添加剂NaOAc 有利于反应的进行。有趣的是,使用NaOAc 作为添加剂,Co(OAc)2和CoCl2表现出类似的催化活性。机理研究表明碘离子的作用最初是通过原位取代氯代烷烃中的氯原子形成活性碘代烷烃,然后在光照作用下进行碘代烷基化合物的羰基化转化[式(23)]。
同年,Wang 等[144]报道了铜和镉盐结合组成的光催化剂(CuBr2、CuCl2和CdI2)体系催化氯代烷烃与CO 的羰化酯化反应。在这些催化剂中,CdI2催化底物生成的目标酯产率和选择性最高,特别是氯代环己烷羰基化的产率和选择性分别达到60%和75%。此外,在CuBr2和CuCl2催化剂体系中加入三正丁胺可以提高酯的产率和选择性。有趣的是单一的CdCl2不进行羰基化反应,但添加NaI·2H2O后,CdCl2的催化活性有所提高(图14)。

图14 非贵金属催化剂催化氯代烷烃与CO的光催化羰基化反应
2022 年,Wang 等[145]首次报道了铑催化未活化烷基氯化物的羰化酯化反应(脂肪族醇或酚均可作为亲核试剂)制备酯类化合物。该反应之所以能发生是因为碘化钠的添加使得氯代物原位转化为活性更高的碘代物中间产物(Monsanto 工艺)。“Rh(acac)(CO)2/DPPP”催化剂体系可一锅法直接制备多种脂类化合物(81 个实例,分离收率最高至95%);当一氯甲烷或者二氯甲烷作为底物时,该催化剂同样可以高效合成多种乙酸酯类化合物。DPPP 的引入不仅可以调控羰基化反应的选择性,还可以调控第一步卤化物交换反应的选择性(图15)。
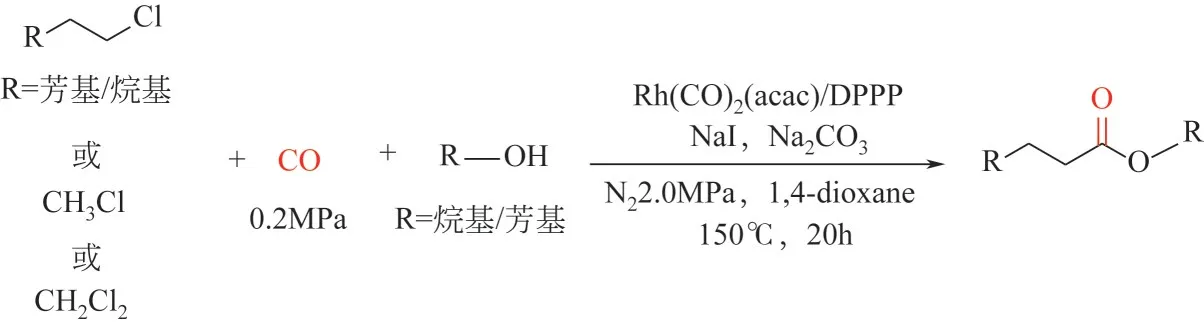
图15 铑催化非活化烷基氯代物的羰化酯化反应
8 结语
氯代物的羰基化反应的产品在大宗化学品、精细化学品、日用品的合成以及药物分子的设计合成中具有广泛的应用。本文综合评述了近年来不同类型的氯代物(芳基氯代物、烯丙基氯代物、苄基氯代物、α-氯代酮、1,1-二氯-1-乙烯基化合物、烷基氯代物)发生羰基化反应的研究进展。该类反应由于底物以及亲核试剂的多样性,可以合成诸多结构多样性的醛、酯、羧酸、酰胺等在工业和药物化学中有广泛应用的化学品;同时合成路线简单,可一步合成所需要的目标产物;目标产物的收率和选择性高,一般均可以达到60%以上;氯代物相对其他卤代物储量丰富且廉价易得,产品具有高附加值,因此经济效益可观。
虽然目前氯代物的羰基化反应研究已经取得了很大进展,但该反应目前仍然存在许多问题亟待解决:①在CO存在下,芳基卤化物与Pd(0)配合物的氧化加成机制尚不明确,酰基钯配合物与亲核试剂的反应机制也尚不清楚;②更深入地了解氯代物与金属配合物的特性,发现更多新的羰基化反应和催化剂体系,诱导芳基氯代物或烷基氯代物的温和羰基化;③均相催化中,氯苯以及烷基氯代物的氢甲酰化反应难度大,目前报道很少;④虽然光催化烷基氯代物的羰基化反应已有报道,但是如何通过加热条件实现过渡金属催化该反应的报道依然很少;⑤虽然一级烷基氯代物的羰基化反应已有报道,但是二级/三级烷基氯代物的羰基化反应(加热条件)尚未有报道。⑥如何在实验室提高目标产物化学/区域选择性的基础上,降低催化剂体系的成本,实现反应规模的放大乃至催化剂的多次循环依然需要探究。同时在工业化生产过程中,如何选择合适的碱添加剂及时中和反应产生的氯化氢也是需要考虑的问题。总之,展望发展更多新型的催化剂体系实现更多类型氯代物的可循环羰基化高效和高选择性转化,实现更多下游高附加值羰基化学品的工业化应用。
猜你喜欢
茶叶通讯(2019年3期)2019-02-16
浙江化工(2019年5期)2019-01-20
中成药(2017年7期)2017-11-22
纺织科学研究(2017年6期)2017-07-03
纺织科学研究(2017年1期)2017-05-17
天然产物研究与开发(2016年11期)2016-06-15
合成化学(2015年2期)2016-01-17
化工进展(2015年6期)2015-11-13
化工进展(2015年3期)2015-11-11
中国塑料(2015年10期)2015-10-14
