
气相色谱质谱法测定蔬菜中氟虫腈及其代谢物与不确定度评定
2023-10-19 02:49林泽珊黄松赵金利谭锦萍黄穗东刘佳
食品工业 2023年9期
林泽珊,黄松,赵金利,谭锦萍,黄穗东,刘佳
广州市食品检验所(广州 511400)
氟虫腈(又名锐劲特),是苯基吡唑类高活性广谱性杀虫剂,被广泛用于蔬菜等农作物中[1-2]。氟虫腈易代谢成氟甲腈、氟虫腈砜和氟虫腈硫醚,其代谢产物毒性更大、持久性更强[3]。我国GB 2763—2021[4]《食品安全国家标准 食品中农药最大残留限量》中规定氟虫腈最大残留限量为0.020 mg/kg,其残留量以氟虫腈、氟虫腈砜、氟虫腈硫醚、氟甲腈之和计,以氟虫腈表示。同时,GB 2763—2021[4]规定蔬菜分析氟虫腈残留应采用SN/T 1982—2007[5]进行测定。但是,该标准只检测氟虫腈原药,并不包含其代谢产物。因此,建立一种蔬菜中同时测定氟虫腈及其代谢物的方法很有必要。
目前,针对氟虫腈及其代谢物的分析方法主要有高效液相色谱串联质谱法[6]、超高效液相色谱串联质谱法[7]、气相色谱串联质谱法[8]。气相色谱串联质谱法具有灵敏度高、特异性强、分析效率高等特点。高霞等[9]研究了气相色谱-负化学源质谱法分析蔬菜中氟虫腈及3种代谢物。郐鹏等[10]针对芹菜单一基质建立了GC-NCI-MS法测定氟虫腈及其代谢物,均未对方法进行不确定度评定。不确定度评定是测量结果准确度的定量表征[11]。在日常检测工作中,当化合物的检测结果位于最大残留限量附近时,对所检测的化合物进行不确定评估,可判断该项目是否合格[12],以提高判定项目的准确度,在食品安全监管和控制方面具有重要意义。刘佳等[13]研究了QuEChERS法前处理结合气相色谱-负化学源质谱分析蔬菜中氟虫腈含量,包含其不确定度评定,但未涉及其代谢产物。
因此,此研究利用填料PSA对样品净化,以基质标准溶液定量,结合气相色谱-负化学源质谱法,建立分析蔬菜中氟虫腈及其代谢物残留的方法。同时,依照JJF 1059.1—2017[14]《测量不确定度评定与表示》、JJF 1135—2005[15]《化学分析测量不确定度评定》、CNAS-CL 06:2019[16]《化学分析中不确定度的评估指南》,对蔬菜中氟虫腈及其代谢物残留进行痕量分析,并对过程产生的不确定度进行评定。该方法前处理简便,在负化学源电离模式下产生碎片离子少,选择性好,灵敏度高,能满足蔬菜中氟虫腈及其代谢物残留量检测要求,为蔬菜中氟虫腈及其代谢物残留量测定的准确性和可靠性提供技术补充。
1 材料与试验方法
1.1 材料与试剂
青瓜、葱、莲藕、胡萝卜、番茄、生菜(广州市某农贸市场)。
氟虫腈、氟虫腈硫醚、氟甲腈、氟虫腈砜(100 μg/mL,农业部环境保护科研监测所);乙腈、正己烷(色谱纯,美国Fisher公司);氯化钠(分析纯,广州化学试剂厂);PSA(上海安谱实验科技股份有限公司);无水硫酸镁(分析纯,广州化学试剂厂)。
1.2 仪器与设备
TSQ 8 000 evo气相色谱-三重四极杆串联质谱仪(NCI离子源,美国Thermo Scientific公司);ME 2002 E电子天平(精密度0.01 g,德国赛多利斯公司);Alluqra X 30 R高速离心机(德国Sigma公司);IKA M53 control涡旋振荡器(德国IKa公司);Organomation N-EVAP 24氮吹仪(美国Organomation公司)。
1.3 试验方法
1.3.1 仪器条件
1) 气相色谱条件:色谱柱-TR-PESTICIDE II毛细管柱(30 m×0.25 mm×0.25 μm);初温:60 ℃,持续1 min,以40 ℃/min升至170 ℃,再以10 ℃/min升至260 ℃,持续3 min;进样口温度:300 ℃;进样量:1 μL;进样方式:不分流;载气:氦气,流量,1.2 mL/min;反应气:甲烷(纯度≥99.999%),1.0 mL/min。
2) 质谱条件:NCI,SRM模式;离子源温度:250℃;质谱参数见表1。
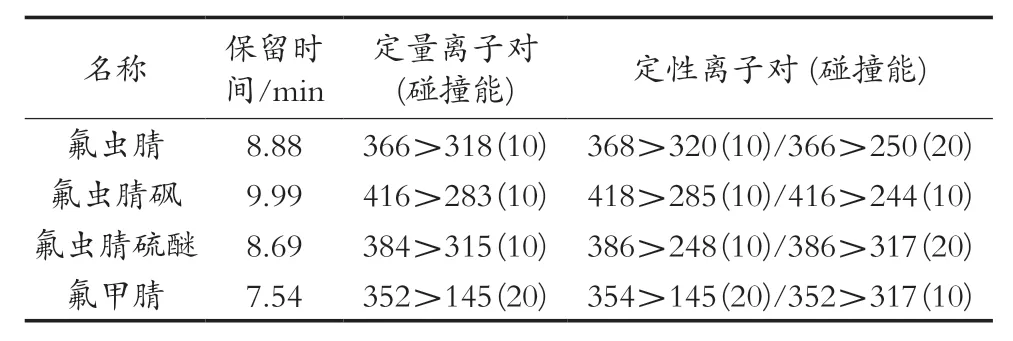
表1 化合物的定性、定量离子对及碰撞能
1.3.2 试验步骤
提取:精确称取10.00 g样品于50 mL透明离心管中,加入20.00 mL乙腈,涡旋提取30 min,加入5 g氯化钠进行盐析,以6 000 r/min离心5 min,得到上清液。
净化:吸取5.00 mL上清液于含有150 mg PSA、900 mg无水硫酸镁净化管中,振荡5 min,按7 000 r/min离心4 min。准确移取1.00 mL至15 mL透明离心管中,于40 ℃氮吹至近干,准确加入1.00 mL正己烷复溶,过0.22 μm有机相滤膜后待测。采用气相色谱-三重四极杆串联质谱仪(NCI源)检测,以保留时间和相对离子对丰度比定性,外标法定量。
基质空白溶液:选取基质样品,按照1.3.2小节试验步骤对其进行前处理,经仪器分析测定,当化合物的定性离子、定量离子的信噪比<3时,则认为该试验样品为空白,试验最后得到的复溶液即为基质空白溶液。
1.3.3 标准溶液配制
混合标准储备液:各吸取1.00 mL 100 μg/mL的氟虫腈、氟甲腈、氟虫腈砜和氟虫腈硫醚标准品于10.00 mL容量瓶,用正己烷定容,得到质量浓度为10.0 μg/mL的混合标准储备液;避光,于0~4 ℃保存,有效期3个月。
混合标准中间液:吸取1.00 mL的混合标准储备液于10.00 mL容量瓶,用正己烷定容,得到质量浓度为1.00 μg/mL的混合标准中间液;避光,于0~4 ℃保存,有效期1个月。
基质混合标准工作液:准确吸取50.0,100.0,200.0,500.0和1 000 μL质量浓度为1 μg/mL的混合标准中间液于同一系列10.00 mL容量瓶中,基质空白溶液定容至刻度,分别得到质量浓度为5.0,10.0,20.0,50.0和100 ng/mL的标准点,供气相色谱-质谱联用仪测定。
1.3.4 回收率测定
选取阴性样品(按照标准分析方法测定确认阴性),精确称出6份10.00 g于50 mL透明离心管中,加入200 μL 1.00 μg/mL的混合标准工作液,即氟虫腈及其代谢物的加标量均为0.02 mg/kg。按照1.3.2小节试验步骤对样品进行处理,测定氟虫腈及其代谢物的残留量,并计算其精密度和回收率。
1.3.5 建立数字模型
采用峰面积外标法定量,菜中氟虫腈、氟虫腈砜、氟虫腈硫醚、氟甲腈残留量按式(1)计算。
式中:X为试样中被测组分残留量,mg/kg;C为试样溶液中仪器检测值,ng/mL:V为试样定容体积,mL;m为试样称取质量,g。
2 结果与分析
2.1 氟虫腈及其代谢物方法的建立
此研究选取了不同类别的蔬菜,以青瓜、葱、莲藕、胡萝卜、番茄、生菜为研究对象,选用乙腈为提取溶剂,经PSA萃取净化,基质标准溶液定量,结合气相色谱-负化学源质谱法,对其方法的检出限、定量限、线性方程、相关系数、回收率、精密度等进行验证,结果见表2和表3。
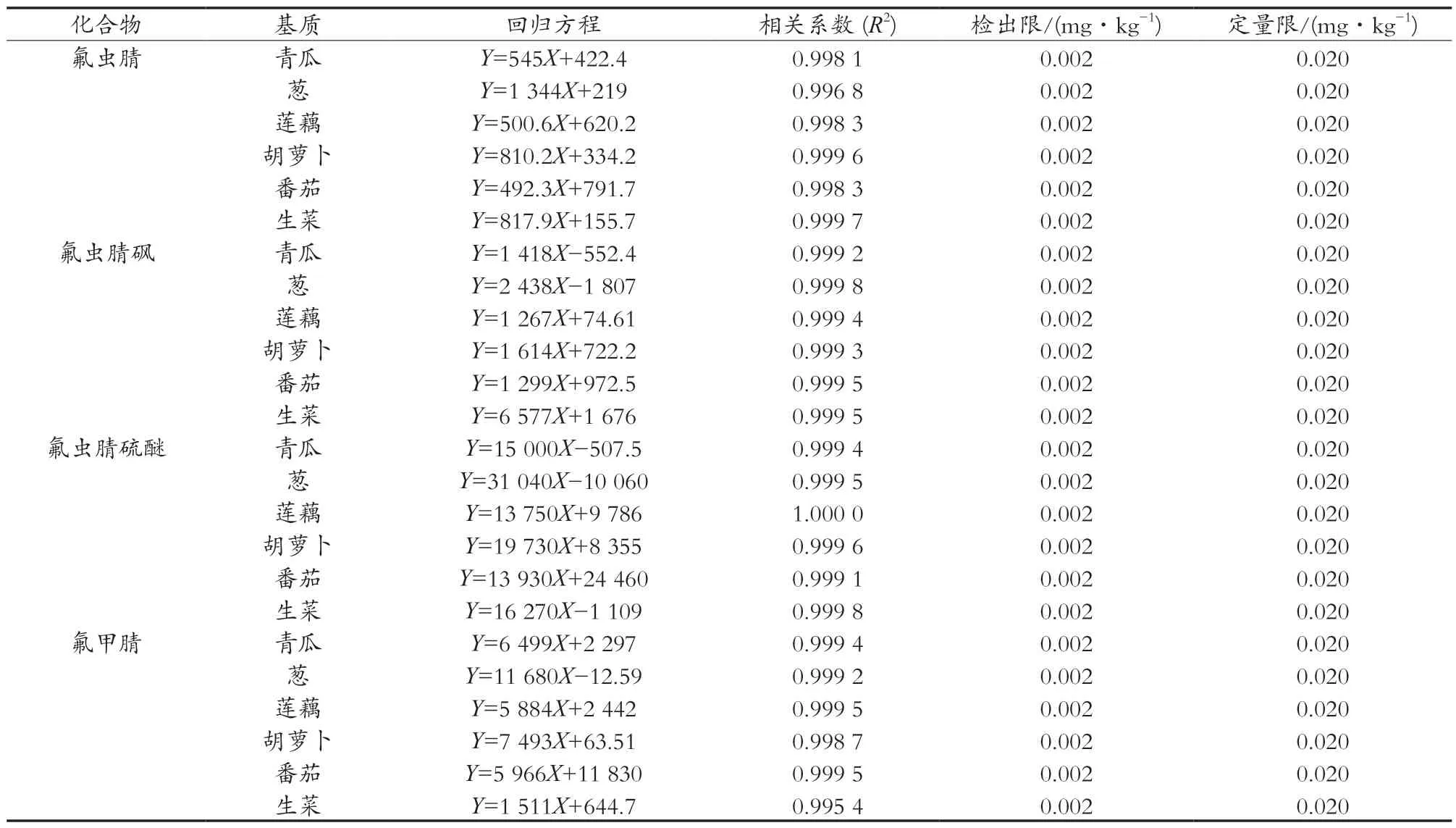
表2 氟虫腈及其代谢物的线性方程、相关系数、检出限、定量限
由表2和表3可看出,氟虫腈及其代谢产物在5~100 ng/mL浓度范围内有良好的线性,其检出限均为0.002 mg/kg。在0.002,0.010和0.020 mg/kg加标水平下,平行测定6次,回收率为86.9%~107.0%,SRSD为0.6%~8.0%,能满足氟虫腈及其代谢物日常测量需求。
2.2 不确定度主要来源
通过分析测量样品过程与数字模型的建立,试验过程引入的不确定度来源主要有称量过程、标准溶液配制过程、前处理过程、样品的重复性和回收率,参考文献[17]将不确定度来源细化,绘制出氟虫腈及其代谢物残留量的不确定度来源鱼骨图,见图1。

图1 氟虫腈及其代谢物残留量的不确定度来源鱼骨图
2.3 不确定度分量评定
2.3.1 称量过程引入的不确定度uRel(m)
试验称样质量为10.00 g(精确至0.01 g),经查阅电子天平检定证书可知,当称量范围在0~50 g时,电子天平最大允许误差为±0.01 g,遵循B类评定方法,取均匀分布,由称量过程引入的相对标准不确定度uRel(m)为0.000 577。
2.3.2 样品前处理过程引入的不确定度uRel(V总)
样品前处理过程的不确定度主要源于移液器以及有机溶剂受环境温度的影响。加入20.00 mL乙腈提取,吸取1 mL净化液转化溶剂,该过程的不确定度主要源于10 mL吸管(A级)和1 mL移液枪以及乙腈受温度变化的影响。按照JJG 646—2006[18]《移液器》检定规程,查阅相关移液器允许误差。已知实验室温度范围20±5 ℃、乙腈的膨胀系数1.37×10-3℃-1,遵循B类评定方法,呈矩形分布,乙腈由温度效应引入的相对不确定度为:0.003 95。
该过程引入的相对合成标准不确定度uRel(10 mL吸管)和uRel(1 mL移液枪)结果见表4。

表4 移液器的相对标准不确定度
试验最后加入1.00 mL正己烷复溶,主要产生的不确定度源于1 mL移液枪。由表4可知,1 mL移液枪引入的相对标准不确定度为0.005 77。按照实验室温度20±5 ℃、正己烷的膨胀系数1.36×10-3℃-1,遵循B类评定方法,按均匀分布,正己烷由温度波动引入的相对不确定度为:
最终样品前处理过程引起的相对标准不确定度uRel(V总)为:
2.3.3 标准物质及配制过程引起的不确定度uRel(标v)
1) 查阅标准物质相应证书信息,收集证书提供的扩展不确定度,参考文献[19]的计算公式,遵循B类评定方法,呈矩形分布计算,标准物质引起的相对标准不确定度uRel(标p)如表5所示。

表5 化合物引入的不确定度uRel(标 p)
2) 制备混合标准中间液引起的不确定度uRel(标V中间液)。由1.3.3小节配制混合标准中间液可知,该过程主要由1 mL移液枪和10 mL容量瓶及正己烷因温度效应引起的不确定度。根据JJG 646—2006[17]《移液器》检定规程和JJG 196—2006[20]《常用玻璃量器检定规程》,查阅相应玻璃器具和移液器允许误差,见表6。遵循B类评定方法,假定呈三角形分布,制备混合标准中间液引入相对标准不确定度urel(标V中间液)为:μrel(标V中间液)=
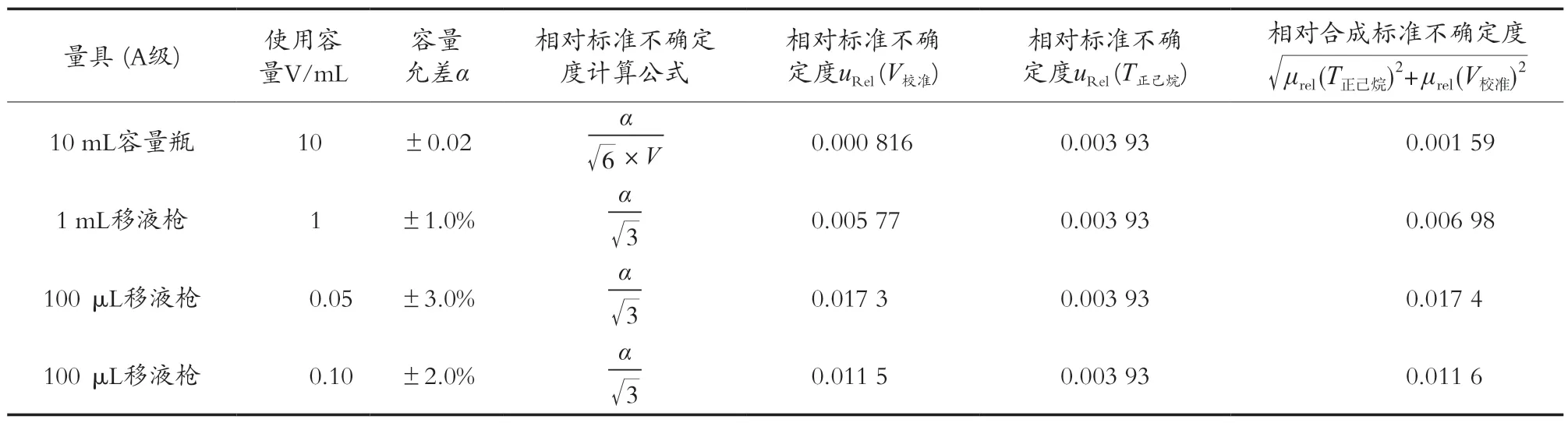
表6 玻璃器具和移液器的相对标准不确定度
3) 曲线配制过程引起的不确定度uRel(标曲)。由1.3.3小节配制基质混合标准工作液可知,该过程共使用了5次10 mL容量瓶、2次1 mL移液枪、4次100 μL移液枪,由表6可得对应相对标准不确定度。因此,制备引入基质混合标准工作液相对不确定度为uRel(标曲):综上所述,氟虫腈及其代谢物基质混合标准品与制备曲线造成的相对不确定度uRel(标v)见表7。
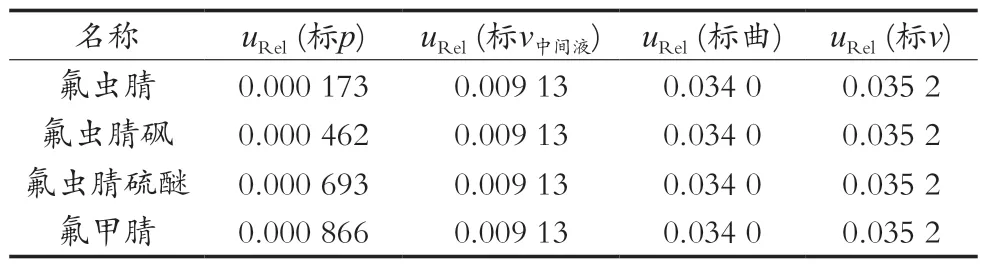
表7 标准化合物及稀释过程引起的不确定度uRel(标v)
2.3.4 样品测量重复性引入的不确定度uRel(S)
选取阴性生菜为试验对象,按1.3.4小节试验步骤对样品进行前处理。遵循A类方法评定中贝塞尔公式[21]计算,氟虫腈及其代谢物的测定结果及相对不确定度度uRel(S)见表8。
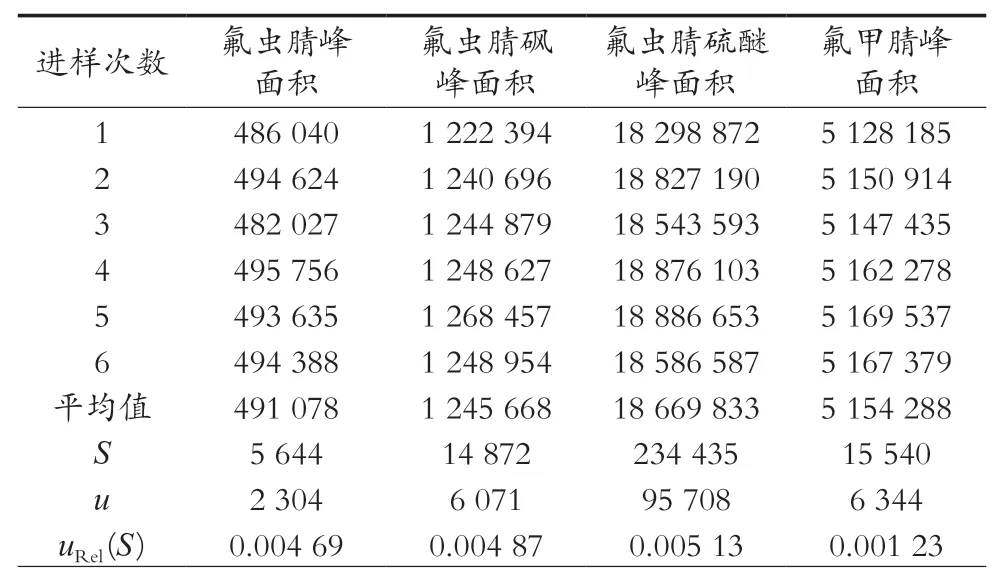
表8 氟虫腈及其代谢物重复测量结果uRel(S)(n=6)
2.3.5 回收率引入的不确定度uRel(Rec)
选择阴性生菜样品,对其进行加标试验,氟虫腈及其代谢物的加标浓度为0.020 mg/kg,平行进行6组试验,各测定结果、回收率以及相对不确定度uRel(Rec)见表9。

表9 氟虫腈及其代谢物的加标回收率引入的不确定度uRel(Rec)(n=6)
2.4 合成标准不确定度
2.5 评定扩展不确定度及报告测量不确定度
依据CNAS-CL 06:2019《化学分析中不确定度的评估指南》[16]理论,假设符合正态分布,置信区间为95%时,选择包含因子k=2,
氟虫腈的扩展不确定度表示如下:
最终测量不确定度报告:(0.020±0.001 5)mg/kg,k=2。
氟虫腈砜扩展不确定度:μ(氟虫腈砜)=μrel(X2)×k×C=0.040 1×2×0.017 mg/kg=0.001 4 mg/kg;最终测量不确定度报告:(0.020±0.001 4)mg/kg,k=2。
氟虫腈硫醚扩展不确定度表示:μ(氟虫腈硫醚)=μrel(X3)×k×C=0.041 5×2×0.018 mg/kg=0.001 5 mg/kg;最终测量不确定度报告:(0.020±0.001 5)mg/kg,k=2。
氟甲腈扩展不确定度表示:μ(氟甲腈)=μrel(X4)×k×C=0.039 0×2×0.018 mg/kg=0.001 4 mg/kg;最终测量不确定度报告:(0.020±0.001 4)mg/kg,k=2。
2.6 不确定度数据分析
通过建立气相色谱-负化学源质谱法检测蔬菜中氟虫腈包含其代谢物的分析方法,并对其进行不确定度评定,不确定度来源分量的贡献见表10。

表10 相对标准不确定度分量
由表10可知,该研究的不确定度主要受标准溶液制备时所产生误差的影响。该过程主要受到移液器、容量瓶、仪器精密度、操作者试验过程中的误差等影响。针对这几个因素,可购买精度更高的测量器具,定期让工程师对仪器进行检定校准,确保仪器稳定性,同时通过提高检测人员的操作水平等,减少对实验结果的影响。
3 结论
此研究建立了气相色谱-负化学源质谱法研究蔬菜中氟虫腈及其代谢物的含量,并对其不确定度评估进行论述。结果表明:该方法快速、简便、重复性好、灵敏度高,适合大批量检测。经对试验过程进行不确定度评定,其来源主要是标准溶液配制过程。因此,在试验中应尽可能避免该过程受不确定因素的影响,减少试验过程产生误差,提高试验数据的准确度。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
食品安全导刊(2021年21期)2021-08-30
应用化工(2021年4期)2021-05-20
理化检验-化学分册(2020年12期)2021-01-26
化工管理(2020年26期)2020-10-09
分析仪器(2019年3期)2019-06-18
山东化工(2019年2期)2019-02-21
农药科学与管理(2019年2期)2019-01-05
环球时报(2017-08-03)2017-08-03
化工科技(2016年6期)2016-06-06
