
基于铁系元素的氨分解制氢催化剂研究进展
2023-12-09 08:17张传坤赵东越孙尚聪宋海涛曹东学
北京化工大学学报(自然科学版) 2023年5期
张传坤 赵东越 孙尚聪 宋海涛 曹东学
(中石化石油化工科学研究院有限公司, 北京 100083)
引 言
随着全球能源需求的持续增长和气候变化问题的日益严峻,传统能源向可再生能源的转型已不可避免。 氢能具有来源多样、用途广泛等优良特性,此外,可再生能源制取的“绿氢”还具有零碳、零污染的特性,因此氢能有望成为解决全球变暖问题的关键[1]。 但氢的沸点极低,其存储和运输较为困难。目前,最常见的氢存储系统采用高压气瓶,但氢气的压缩需要耗费大量的能量[2],而氢气的液化同样面临能耗及氢气蒸发等问题,因此氢作为能量载体的发展和利用受到了限制[3]。
氨作为一种储氢材料,具有多种优势[4]。 首先,氨具有较高的含氢量(质量分数为17.6%)和能量密度(3 000 W·h/kg),其在分解过程中不会产生碳排放[5];其次,氨在常压下的沸点为-33.5 ℃,易于液化,其存储和运输也较为方便;最后,合成氨的历史已有100 多年,现有的工业体系较为完整和成熟。 这些优势都有利于氨的规模化利用,特别是在现场制氢中,氨被认为具有广阔的发展前景[6]。 但是,氨分解的温度较高,分解过程所需的能耗较大,这导致氨作为储氢材料未能得到广泛应用[7]。 因此,能够在较低温度下高效低成本催化氨分解制氢的催化剂日益受到研究者的重视。
目前,常应用于氨分解催化剂的金属有Ru、Ni、Fe、Co 等。 贵金属Ru 在氨分解反应中表现出的活性最高[8],因此其研究也最多,但是Ru 高昂的价格限制了它的大规模应用[9]。 铁系元素(Ni、Fe、Co)以成本优势引起了人们的关注,但其活性仍需要进一步提高。 本文以提高铁系元素氨分解催化剂的活性为出发点,从氨分解基本原理、催化剂的活性组分、载体和助剂4 个方面,对铁系元素氨分解催化剂进行了总结和分析,并对其未来的发展趋势进行了展望。
1 氨分解基本原理
氨分解反应是体积增大的吸热反应(式(1)),升高温度、降低压力有利于提高反应的平衡转化率。在400 ℃以上时,氨分解的平衡转化率可以超过99.1%,因此从热力学角度看,在400 ℃以上时,氨可以在催化剂的作用下完全分解[10]。
催化剂催化氨分解是一个逐步脱氢的过程[8]:首先氨分子吸附在催化剂表面,之后表面的氨分子逐步脱去H 原子,最后表面的N 原子和H 原子重组脱附生成N2和H2(式(2) ~(9))。
人们对氨分解反应动力学进行了较多的研究,但并没有统一的动力学模型可以对所有条件下的氨分解过程进行描述。 目前,采用较多的模型是Temkin-Pyzhev 动力学方程(式(10)),该方程立足于两个基本假设:(1)氮分子的表面脱附为速控步骤;(2)脱附速率与表面覆盖率呈指数关系[11]。
式中:rNH3为氨分解反应速率;R为气体常数;T为反应温度(热力学温度);k0和Eap分别为指前因子和活化能;pNH3和pH2分别为NH3和H2的分压;α和β分别为NH3和H2的反应级数,其中α>0,β<0,这说明增大NH3分压、降低H2分压可以增加氨分解的速率,而N2分压对反应速率没有影响。
目前,对铁系元素催化氨分解的限速步骤仍然存有争议。 Arabczyk 等[12]对Fe 催化氨分解的研究表明,Fe 催化氨分解过程中同时存在Fe 的氮化反应,而Fe 的氮化物会降低氨分解的反应速率,这说明Fe 表面N 的重组脱附是限速步骤。 Seyfeli 等[13]对微波反应器中Co 催化氨分解过程的研究表明,微波可以促进Co 表面N 的重组脱附,提高催化剂的活性,这表明Co 催化氨分解的限速步骤也是N的重组脱附。 数坂昭夫等[14]以15N 为示踪剂,计算了氨分解过程中各产物的比例,结果表明在较低温度下,Fe 催化氨分解的限速步骤是N 的重组脱附,但高温时氨的脱氢是限速步骤。 Duan 等[15]利用密度泛函理论研究了Ni(110)面催化氨分解的过程,结果显示Ni 表面N 原子的脱附过程所需的能量最高,因此N 的重组脱附是限速步骤。 Takahashi等[16]将NH3的脱氢反应视为一步反应,并对氨分解各步反应的动力学过程进行了研究,结果表明,NH3在Ni 表面的脱氢较困难,因此NH3脱氢是Ni 催化氨分解的限速步骤。 以上研究表明,铁系元素催化氨分解的限速步骤主要是N 的重组脱附步骤或NH3的脱氢步骤,但人们并没有统一的认识,这种争议的存在可能意味着对铁系元素催化氨分解限速步骤的研究需要根据具体情况进行具体分析。
2 催化剂活性组分
3 种铁系元素同属于第VIII 族元素,但在不同因素的影响下,其催化活性存在差异。 Ganley 等[17]的研究表明,铁系元素表面N 的脱附过程是其催化氨分解的限速步骤。 铁系元素中Ni 表面N 的脱附速率最快,因而活性也最高,三者的活性大小顺序为:Ni >Co >Fe。 铁系元素与N 的结合能是造成其活性差异的重要因素,金属与N 最适宜的结合能为561 kJ/mol,铁系元素与N 的结合能越接近该值,活性越高[18]。 Fe、Co、Ni 这3 种金属与N 的结合能分别为619.5、523.2、510.7 kJ/mol[19],其中Fe 与N 的结合能过高,导致形成FeNx,因此其活性最低。 根据结合能的大小,3 种金属的活性顺序为:Co >Ni >Fe。 Gu 等[20]的研究表明,铁系元素的抗烧结能力也会导致这3 种金属的活性差异。 在600 ℃时,Co和Ni 容易发生烧结团聚,因此活性较低,而Fe 由于形成了Fe2N,保持了较高的活性,此时活性大小顺序为:Fe >Co >Ni。 此外,铁系元素活性的差异还与金属的存在形式有关。 当铁系元素以金属形式存在时,其活性较高,活性大小顺序为:Fe >Co >Ni,但随着反应的进行,Fe 和Co 被完全氮化,而Ni 表面发生氮化,导致三者的活性均显著降低。 在不同因素的影响下,铁系元素在不同的催化体系中表现出了不同的催化活性,但更多的研究表明,Ni 可能比Fe 和Co 具有更高的催化活性,而Fe 的活性则较低(图1)[18]。
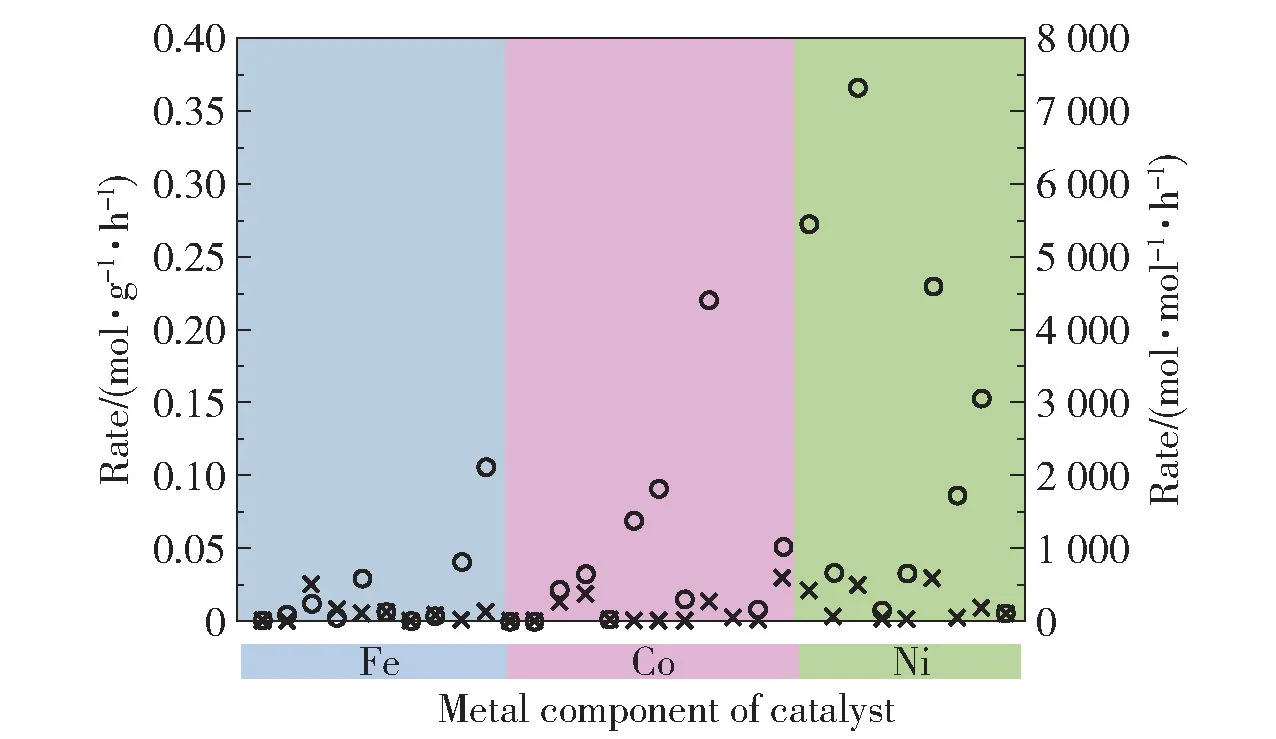
图1 500 ℃时Fe、Co 和Ni 催化剂的氢气生成速率汇总[18]Fig.1 Summary of hydrogen formation rates for reported iron,cobalt and nickel catalysts at 500 ℃[18]
根据活性组分中金属组分数的不同,铁系元素氨分解催化剂主要分为单金属催化剂和双金属催化剂。 与合成氨反应相同,铁系元素催化氨分解也是结构敏感型反应[21-22],晶粒尺寸和晶面对催化剂的活性有较大影响。 为了降低成本、提高活性,人们对铁系元素双金属催化剂进行了较多的研究。 本文从晶粒尺寸、晶面和双金属组成这3 个方面对铁系元素催化氨分解的研究进行介绍。
2.1 晶粒尺寸
较小的晶粒尺寸可以提高催化剂的比表面积和活性表面积,进而提高催化剂的活性。 但催化剂的活性表面积与晶粒尺寸并不存在线性关系。 Pelka等[23]在研究熔铁催化剂的氨分解性能时发现,在28 ~40 nm 范围内,随着晶粒尺寸的减小,催化剂的比表面积增加,但活性表面积的增加逐渐放缓。 原因是比表面积的增加使熔铁催化剂单位表面上助剂钾(K)的浓度降低,导致由K 构成的活性位点(图2)的数量下降[24]。 活性表面积的增加逐渐放缓意味着催化剂的晶粒尺寸并不是越小越好。Zhang 等[25]对Ni0催化氨分解过程的研究表明,过大或过小的晶粒尺寸都会导致催化剂活性降低,当Ni0晶粒尺寸约为2.2 nm 时,催化剂的转换频率(TOF)达到最大值29.5 s-1,此时催化剂的活性最高。 原因是Ni0催化氨分解的活性与B5 活性位点有关,在该晶粒尺寸下B5 活性位点出现的概率最大。 Torrente-Murciano 等[26]对负载于不同碳材料上Co 催化剂的研究表明,Co 的最佳晶粒尺寸在2 nm以内,原因是在该晶粒尺寸的范围内,具有更高活性的C7 位点数量更多。 需要指出的是,在不同的催化体系中,同种元素的最佳晶粒尺寸并不相同。Duan 等[22]的研究表明,Ni 的最佳晶粒尺寸为8.9 ~11.0 nm,当晶粒过小时,容易被表面N 阻塞的台阶位点数量过多,导致催化剂活性降低;而晶粒过大时,台阶位点数量过少,不利于NH3的脱氢,也会导致催化剂活性降低,该研究结果与Zhang 等[25]认为的最佳晶粒尺寸(2.2 nm)不同。
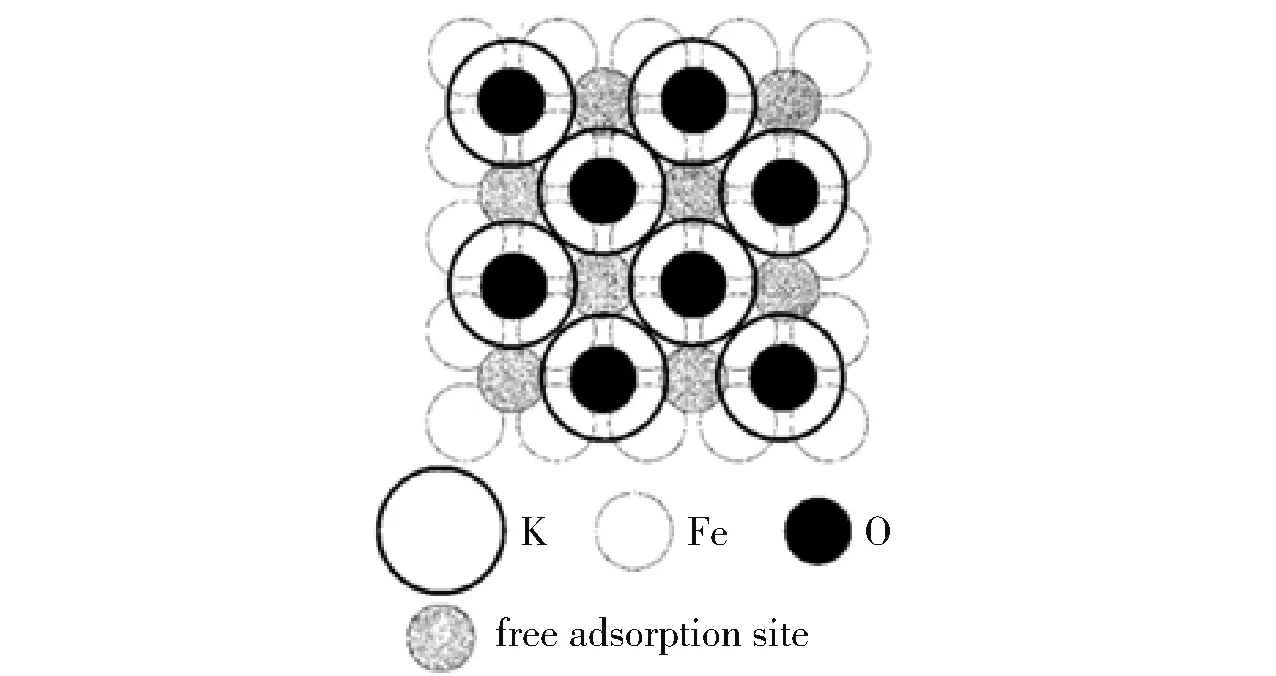
图2 被K 和O 覆盖的Fe(100)面的俯视图[24]Fig.2 Top view on the Fe(100) surface covered with potassium and oxygen[24]
以上研究表明,铁系元素氨分解催化剂存在最佳的晶粒尺寸,在该晶粒尺寸下催化剂的活性最高,但在不同的催化体系中,催化剂的活性位点和最佳晶粒尺寸并不相同。 催化剂的晶粒尺寸会对催化剂的活性位点产生直接影响,确定不同催化剂的活性位点并调控晶粒尺寸以提高活性位点的数量或比例是提高催化剂活性的重要方法。
2.2 晶面
对铁系元素不同晶面的研究主要按照吸附-活化机理进行。 Duan 等[27]对Fe(110)、Co(111)和Ni(111)晶面的研究表明,这3 种晶面上中间产物的最稳定吸附构型存在差异(图3),但3 种晶面上N的重组脱附活化能都比NH3的脱氢活化能高,因此N 的重组脱附活化能是判断不同晶面活性的关键因素。 Fe(110)面的N 重组脱附活化能最高,使表面N 不易脱附,因而活性较低,而Co(111)和Ni(111)面的N 重组脱附活化能较低,因此活性较高。 Ji等[28]研究了Fe 的4 种晶面上N 重组脱附活化能,结果表明,台阶面(Fe(111)和Fe(211)面)上N 的重组脱附活化能比平台面(Fe(100)和Fe(110)面)低,因此活性较高,原因可能是台阶面上存在C7 位点(图4),而C7 位点可以降低N 的重组脱附能。Stolbov 等[29]的研究表明,氨分解的活性不仅与晶面上N 原子重组脱附的难易程度有关,还与NH3的吸附能(Ead)和NH3脱去第一个氢原子的解离能(E1)的能量对比有关:当E1大于Ead时,NH3倾向于脱附而不是脱氢;当E1小于或接近于Ead时,NH3才倾向于脱氢分解。
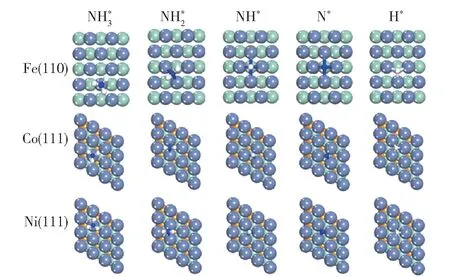
图3 Fe(110)、Co(111)和Ni(111)晶面上NHx(x =0 ~3)和H 的最稳定吸附构型[27]Fig.3 The most stable adsorption configurations of NHx(x=0 -3) and H on Fe(110), Co(111) and Ni(111)surfaces[27]
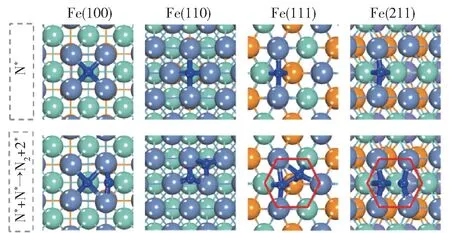
图4 Fe(100)、Fe(110)、Fe(111)和Fe(211)晶面上N的最稳定吸附构型和N 的重组脱附过渡态[28]Fig.4 The most stable adsorption configuration of N and the transition states of the recombinative desorption of N on Fe(100), Fe(110), Fe(111), and Fe(211)surfaces[28]
除了对不同晶面上的氨分解活性进行研究外,Ji 等[30]还研究了负载于碳纳米纤维(CNFs)上的Ni纳米颗粒,结果表明,Ni 催化氨分解的活性与Ni 纳米颗粒的形貌存在相关性:具有多面体形貌的Ni 纳米颗粒比梨形Ni 纳米颗粒具有更高的活性,其中多面体形貌最明显的Ni-CNF3(图5)的活性最高,原因是该种形貌的Ni 纳米颗粒的Ni(110)和Ni(111)面的比例最佳,Ni(110)面有利于氨脱除第一个氢原子,Ni(111)面则有利于其他步骤。
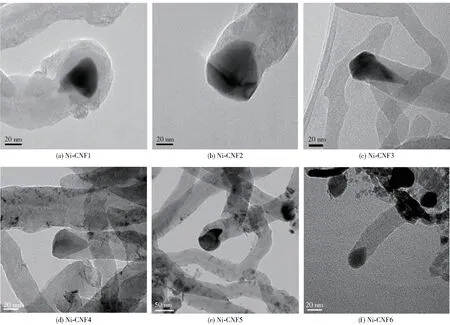
图5 Ni-CNFs 催化剂的高分辨透射电子显微镜(HRTEM)图像[30]Fig.5 HRTEM images of Ni-CNFs catalysts[30]
以上研究表明,铁系元素不同晶面上原子排列方式的不同使氨分解过程中间产物的最稳定构型存在差异,进而影响反应步骤的活化能,导致不同晶面具有不同的反应活性,因此晶面的活性与晶面上存在的活性位点有关。 通过改变催化剂纳米颗粒的形貌可以对晶面的数量和比例进行调控,适当比例的不同晶面可以发挥协同作用,从而进一步提高催化剂的活性。
2.3 双金属组成
一般而言,双金属催化剂具有与单金属催化剂不同的性质。 在某些组合下,双金属催化剂具有比单金属催化剂更高的活性。 金属与N 的结合能被认为是评价金属催化氨分解活性的关键因素[19]。适宜的金属和N 的结合能有利于提高催化剂的活性,结合能过高和过低都会导致活性降低(图6)[31]。Ru 具有最适宜的结合能,因而活性最高。 利用元素周期表插值法,将较低和较高氮结合能的金属按照一定比例进行组合,可能会得到具有较适宜结合能的双金属催化剂,从而提高催化剂的活性[32]。 表1 为文献报道的铁系元素双金属催化剂的组成及催化剂性能比较。 需要指出的是,双金属催化剂的活性不仅受到金属和N 结合能的影响,还受到活性组分周围的环境、金属在表面的偏析和氮的诱导重组等因素[19,32]的影响,此时利用金属与N 的结合能预测的双金属催化剂的活性与实际情况会存在偏差[19]。
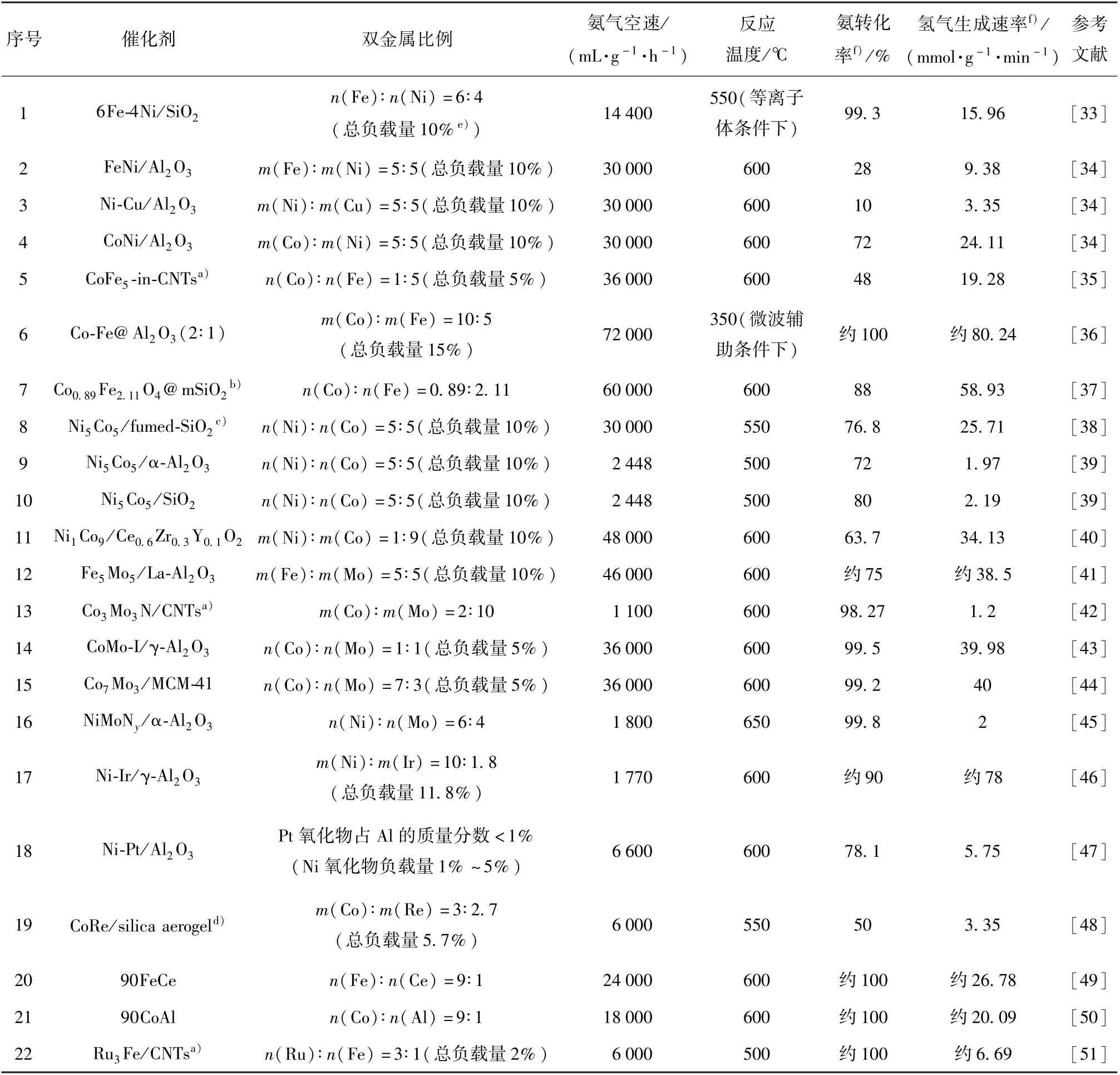
表1 铁系元素双金属催化剂的组成及催化性能比较Table 1 Composition and catalytic performance comparison of iron group element-based bimetallic catalysts
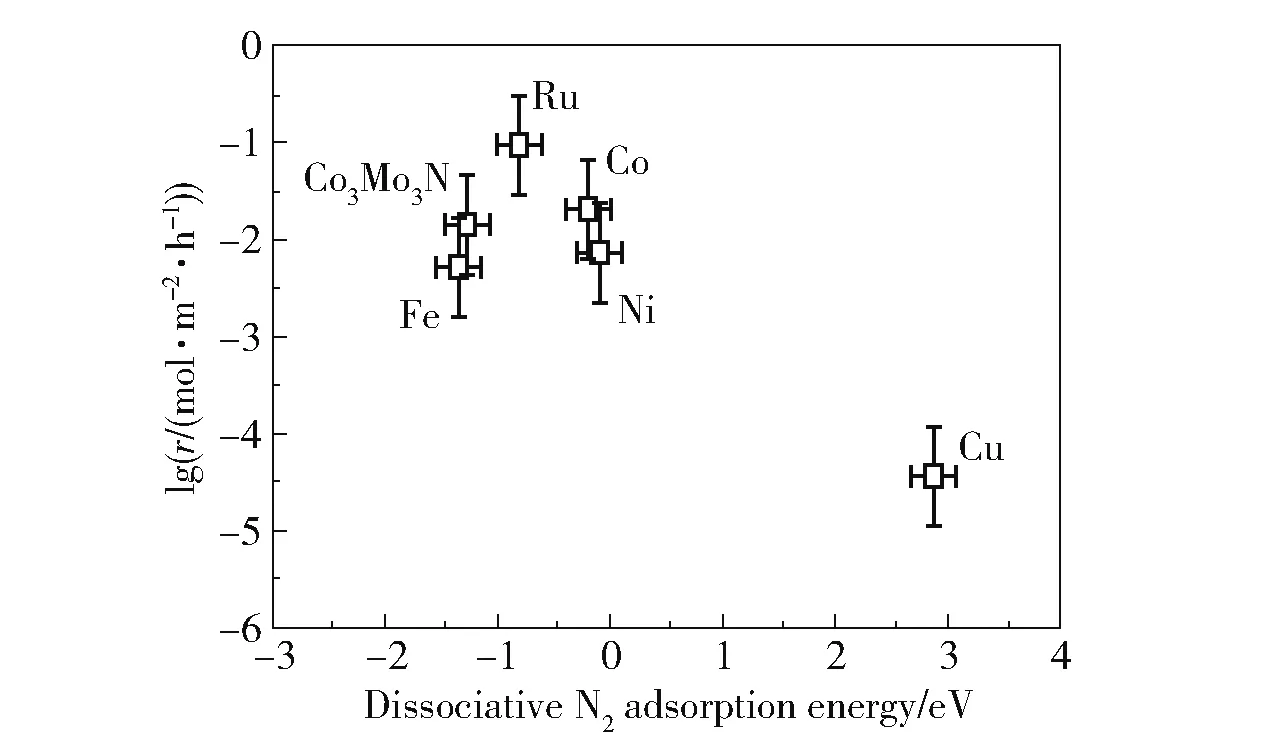
图6 在773 K、100 kPa、V(H2)∶V(N2) =3∶1、20% NH3的条件下不同催化剂的氨分解反应速率[31]Fig.6 Ammonia decomposition reaction rate over different catalysts at 773 K,100 kPa,V(H2)∶V(N2) =3∶1,and 20% NH3[31]
根据双金属的分布构型,双金属催化剂可以分为单层双金属催化剂和双金属合金催化剂[52]。 其中,单层双金属催化剂的附加金属附着于主金属表层或亚表层,金属的表面偏析和核壳型结构都可归于此类,见图7(a)[53];双金属合金催化剂的两种金属都存在于表面和体相,见图7(b)[53]。

图7 单层双金属和双金属合金的结构示意图[53]Fig.7 Schematic representation of the structure of a bimetallic monolayer and a bimetallic alloy[53]
2.3.1 单层双金属催化剂
单层双金属催化剂的N 结合能可以通过理论计算得到,从而根据N 结合能预测双金属催化剂的活性。 Hansgen 团队[19,54]利用密度泛函理论和微动力学模型,对多种单层双金属催化剂的N 结合能进行了计算,结果表明,负载于金属Pt 上具有表面构型的Ni-Pt-Pt(111)单层双金属催化剂的氨分解活性最高(N 结合能最适宜,约为560.9 kJ/mol)。 实验结果也表明,这种催化剂在较低的脱氮温度下,具有比Ru 更高的活性。 考虑到单层双金属结构的稳定性,当Co 或Fe 能与Pt 构成核壳型结构时,催化剂表现出较高的活性,但当Cu 与Pt 组合时,氨分解活性较低。
核壳型单层双金属催化剂催化氨分解的活性也可以通过密度泛函理论进行预测。 Chen 等[55]利用密度泛函理论预测了具有核壳型结构的M@ Ni(M=Fe、Ru、Ir,M 为核心原子,Ni 为壳层原子)催化剂的活性(图8),结果表明,3 种核壳型催化剂中,总体上Ru@Ni 具有较低的位能线,因此其活性较高。 构成单层双金属催化剂的两种金属之间的电子转移是其比单一金属具有更高活性的原因[56]。Zhong 等[57]对具有亚表面构型的Pt-Ni-Pt(111)催化剂的研究结果表明,在Pt(111)亚表面层沉积Ni后,Pt 4f 电子结合能升高,Ni 2p 电子结合能降低,发生了从Pt 向Ni 的电子转移,而这种电子的再分配降低了金属和N 的结合能,促进了表面N 的脱附,从而提高了催化剂的活性。

图8 Fe@Ni、Ru@Ni、Ir@Ni 纳米颗粒和Ru(0001)面上NH3分解脱氢过程和N2脱附过程的势能比较[55]Fig.8 Potential energy comparison of the NH3 decomposition process and the N2 desorption process on Fe@Ni,Ru@Ni,Ir@Ni nanoparticles and a Ru(0001) surface[55]
2.3.2 双金属合金催化剂
在双金属合金催化剂的研究中,不同金属种类的组合是一个重要的研究方向,合适的双金属组合可以产生协同作用,提高催化活性。 Fu 等[34]对MNi/Al2O3(M=Co,Fe,Cu)双金属催化剂的研究结果表明,3 种组合中M 金属都与Ni 形成了合金,但FeNi 合金和CuNi 合金提高了N 结合能,降低了氨分解的活性,而CoNi 合金降低了N 结合能(同时减小了粒径尺寸),提高了催化活性。 在650 ℃时,Co-Ni/Al2O3催化氨分解的转化率超过90%,比在相同条件下Ni/Al2O3催化氨分解的转化率提高了约50%。 Mo 则可以与铁系元素形成三元氮化物[58](Co3Mo3N、Ni2Mo3N、Ni3Mo3N、Fe3Mo3N 等)而促进氨的分解。 Ji 等[43]对CoMo/γ-Al2O3的研究结果表明,采用Co(en)3MoO4(en 为乙二胺)作为前驱体制备的CoMo 双金属催化剂比采用Co(NO3)2和(NH4)6Mo7O24作为前驱体制备的CoMo 双金属催化剂具有更高的活性,原因是采用Co(en)3MoO4制备的催化剂形成了更多的Co3Mo3N,Mo 促进了Co 的分散,Co 则降低了Mo 表面N 的脱附能,因此Co3Mo3N 含量越大,氨分解活性越高。 Lorenzut 等[41]对Fe-Mo/Al2O3催化剂的研究结果表明,Fe(Mo)3N1.24也具有良好的氨分解活性。
此外,双金属合金催化剂中双金属的比例也会影响催化剂的活性。 在一定比例下,双金属之间的协同作用最强,催化剂活性最高。 Yi 等[59]在等离子体辅助条件下,对FeNi/f-SiO2(f-SiO2为气相SiO2)双金属催化剂的研究结果表明,随着Ni 含量的增加,催化剂的活性先增加后降低(如图9 所示,发射光谱(OES)强度越低,催化剂表面上吸附的激发态物种越多)。 Fe 可以与N 原子相互作用,而Ni与H 原子相互作用,当Fe 和Ni 的物质的量比为6∶4时,FeNi 合金具有最多的吸附位点,有利于激发态物种的吸附。 而等离子体不仅促进了激发态物种的产生,还促进了表面物种的脱附,因此这种催化剂在等离子体辅助下表现出良好的活性,在500 ℃时,氨的转化率可以达到99.9%,H2生成速率达到16 mmol/(g·min)。

图9 等离子体催化NH3分解反应中564.2 nm 的OES 强度与催化剂TOF 的关系[59]Fig.9 The relationship between the OES intensity at 564.2 nm and TOF of catalysts in plasma-catalytic NH3 decomposition[59]
除了铁系元素与非贵金属外,铁系元素与贵金属组成的双金属也得到了研究,这种双金属催化剂表现出较好的活性。 Chen 等[51]采用液相还原法制备了Ru-Fe/CNTs 双金属催化剂,稳定性测试结果表明,起初双金属催化剂具有与Ru/CNTs相似的活性,但60 h 后,Ru3Fe/CNTs(Ru 与Fe 物质的量比为3 ∶1)的活性下降了16.8%,而Ru/CNTs 的活性下降了27.5%,说明双金属催化剂在具有较高活性的同时也保持了较好的稳定性。 原因是双金属催化剂上表面N 原子的覆盖率较低,防止了B5 位点的阻塞,并且N 在合金表面更容易脱附。 Yang 等[60]介绍了一种利用沸石-咪唑酯骨架制备Ru clusters@N-C 和Ru-Co clusters@N-C 的方法(图10),Ru 纳米团簇表面低配位和不饱和活性位点促进了氨的脱附和分解,而Co 的合金化效应改变了Ru 纳米团簇的局部结构,使其具有更高的活性,在798 K 时,Co 的加入使Ru clusters@N-C的活性提高了45 倍。
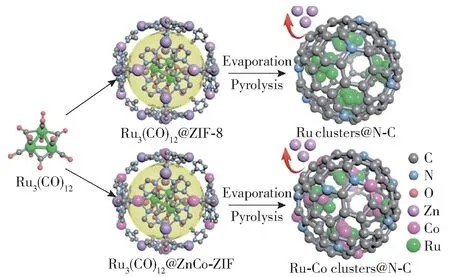
图10 Ru clusters@N-C 和Ru-Co clusters@N-C 的制备工艺[60]Fig.10 Preparation processes of the Ru clusters@N-C and Ru-Co clusters@N-C[60]
根据以上研究可以发现,单层双金属催化剂中两种金属之间的电子转移是其比单金属活性更高的原因,而且其活性可以通过密度泛函理论等方法进行预测,这为铁系元素双金属催化剂的开发提供了非经验性的解决方案,但单层双金属催化剂在具体实验条件下的稳定性仍需进一步研究[61]。 双金属合金催化剂中合适的金属种类及比例是影响其活性的关键因素,而铁系元素与贵金属组成的双金属合金可能比铁系元素与非贵金属组成的双金属合金具有更高的活性[52]。 从平衡成本和活性的角度考虑,铁系元素与Ru 等贵金属组成的双金属催化剂可能是一个十分具有前景的研究方向[62]。
3 载体
目前,应用于铁系元素氨分解催化剂的载体较多,不同载体的特性对催化剂性能的影响也不相同。一般来说,这些载体主要与导电性、碱性和金属分散度等性质有关。 表2 为铁系元素氨分解催化剂的载体及催化性能比较。

表2 铁系元素氨分解催化剂的载体及催化性能比较Table 2 Supports and catalytic performance comparison of iron group element-based catalysts for ammonia decomposition
3.1 碳材料载体
常用作氨分解催化剂载体的碳材料包括碳纳米管、活性炭(AC)、石墨烯(GO)等,碳材料载体的特性对催化剂的活性有重要影响。
碳材料优良的导电性有利于提高催化剂的活性。 Zhang 等[95]对4 种不同类型的碳材料载体进行了研究,发现在400 ~500 ℃的范围内,Co 负载量为5%时,催化剂的活性大小顺序为:Co/MWCNTs >Co/AC >Co/rGO >Co/SWCNTs(单层碳纳米管)。当NH3空速为6 000 mL/(g·h)、温度为500 ℃时,Co/MWCNTs 催化NH3分解的转化率可以达到约60%,MWCNTs 优异的导电性有利于电子的传导,从而促进了N 的脱附,提高了催化剂的活性。Zhang 等[69]对Ni/MWCNTs 和Ni/AC 的研究同样表明,MWCNTs 的高导电性使Ni/MWCNTs 表现出比Ni/AC 更高的活性。 Meng 等[71]对Ni/rGO 的研究结果表明,石墨烯的高导电性提高了催化剂的活性,在700 ℃时,10%镍负载的Ni/rGO 催化氨分解的转化率达到81.9%,H2生成速率达到27.4 mmol/(g·min)。
碳材料的石墨化程度也会影响催化剂的性能,但在不同催化剂体系下,其对催化剂活性的影响不同。 Torrente-Murciano 等[26]对Ax-21 和MSC-30 碳载体负载的Co 催化剂的研究表明,在具有相似粒径尺寸的情况下,Co/Ax-21 比Co/MSC-30 具有更高的活性,原因是MSC-30 较高的石墨化程度提高了载体的供电子能力,对Co 催化氨分解有不利影响。与此相反,Duan 等[96]在对Fe-CNFs/mica 的研究中认为,高石墨化程度有利于保持CNFs 的稳定性,从而提高催化剂的活性。 Akarçay 等[97]对Fe@C 的研究表明,高石墨化程度的碳载体有利于电子传递,促进了电子向Fe 的转移,从而提高催化剂的活性。 阳卫军等[98]在研究Fe/AC 催化剂时发现,载体的石墨化也有利于提高催化剂的活性。
一般而言,碳材料的导电性受到碳材料石墨化程度的影响。 石墨中碳原子的电子轨道以sp2形式杂化,未参与杂化的电子可自由移动,这是石墨能够导电的原因。 因此,石墨化程度越高的碳载体,其导电性也越好。 MWCNTs 的石墨化程度较高,因此其导电性也较好。 碳材料作为铁系元素氨分解催化剂的载体时,虽然较高的石墨化程度有利于提高载体的导电性,有利于电子向负载金属转移,促进表面N的脱附[95],从而提高催化剂的活性,但是石墨化程度过高使碳材料的结构缺陷较少,不利于金属颗粒沉积,从而降低催化剂的分散程度和催化活性。 此外,碳材料的导电性还与碳材料的孔隙结构有关,具有发达孔隙结构的碳材料,其导电性较差。 活性炭的孔隙结构发达、石墨化程度较低,导致其导电性较差[99]。 最后,碳材料的导电性还与负载金属有关,负载金属可以提高碳载体的导电性。 Liang 等[100]的研究发现,N 掺杂的CNTs(NCNTs)经轻度氧化后生成的表面-COO 物种有利于纳米晶体的成核和生长,而负载Co 后生成的CoO 和碳载体之间存在的强耦合作用提高了载体的导电性能。 Liu 等[101]对Ru 修饰的双层碳纳米管的研究结果表明,Ru 负载于CNTs 上时,电子从Ru 向CNTs 转移,降低了CNTs 的功函数,提高了CNTs 的导电性。
上述研究表明,碳载体的高导电性是提高铁系元素氨分解催化剂活性的有利因素,MWCNTs 以其较高的导电性成为一种有效的碳载体。 石墨化程度对催化剂性能的影响仍有争议,值得进一步研究。需要指出的是,在高温(500 ℃以上)、H2气氛下,碳材料易发生甲烷化反应,且CNTs 的价格较高,不利于大规模应用[9]。
3.2 Al2O3载体
Al2O3价格便宜,耐热性高,比表面积大,是一种应用十分广泛的催化剂载体。 Al2O3载体较大的比表面积有利于提高金属的分散度、减小晶粒尺寸,进而提高催化剂活性。 Gu 等[102]采用一锅法合成了负载于多孔Al2O3上的高分散度的Ni@ Al2O3催化剂(图11),活性测试结果表明,在600 ℃、Ni 和Al物质的量比为1∶3、NH3空速为24 000 mL/(g·h)的条件下,NH3转化率可以达到93.9%,H2生成速率达到25.1 mmol/(g·min)。 Ni 与多孔Al2O3载体强烈的相互作用促进了Ni 的分散,提高了Ni 的抗烧结能力,分散性良好的小颗粒Ni 是该催化剂具有较高活性的原因。

图11 尺寸均匀的小颗粒Ni 负载于多孔Al2O3上[102]Fig.11 Loading of small Ni particles with uniform size on porous Al2O3[102]
Al2O3载体可以提高负载金属的分散度,而对Al2O3载体改性可以进一步提高催化剂的活性。Verwey[103]指出,γ-Al2O3的晶体结构与含有阳离子空位的缺陷尖晶石结构类似,这种空位有利于利用异价阳离子对γ-Al2O3进行改性。 Henpraserttae等[73]采用Zr 对Al2O3进行掺杂,制备了Zr-doped Al2O3载体,之后对其负载的Ni 催化氨分解的性能进行了研究,结果表明,Ni/Zr-doped Al2O3的活性比Ni/γ-Al2O3有明显提高(图12)。 在550 ℃、7 500 mL/(g·h)的氨气空速下,Ni/Zr-doped Al2O3催化氨分解的转化率达到79.8%,H2生成速率达到5.5 mmol/(g·min);而在相同条件下,Ni/γ-Al2O3催化氨分解的转化率只有56.8%,H2生成速率只有4.0 mmol/(g·min)。 原因是Zr 的掺杂提高了Ni 的分散度,增大了载体的比表面积和碱性,进而提高了催化剂的活性。 Lorenzut 等[41]对Fe-Mo/Al2O3的研究结果表明,当载体为La 改性的Al2O3时,催化剂的活性得到了显著提高,原因是La 的加入提高了载体的碱性。

图12 Ni/Zr-doped Al2 O3 和Ni/γ-Al2 O3 在不同温度下催化氨分解的转化率[73]Fig.12 Conversion of ammonia over Ni/Zr-doped Al2O3 and Ni/γ-Al2O3 at different temperatures[73]
Vacharapong 等[104]研究了在Ce-doped Al2O3载体的制备过程中磁场诱导效应对Ni 基催化剂性能的影响,发现在凝胶法制备载体的过程中对凝胶施加相同极性的磁场(在凝胶两侧放置相同极性的磁极,即同为N 磁极或同为S 磁极),可以使Ce 在Al2O3骨架上分布更均匀,从而提高了载体的比表面积、Ni 的分散度和Lewis 碱性位点(图13)的数量,进而提高了催化剂的活性。 在500 ℃、75 000 mL/(g·h)的氨气空速下,在施加相同极性的磁场时制备的Ni/Ce-doped Al2O3催化氨分解的转化率达到约51%,H2生成速率达到约4.3 mmol/(g·min);而未施加磁场和施加相异极性的磁场(在凝胶两侧分别放置不同极性的磁极,一侧为N 磁极,另一侧为S 磁极)时,所制备的Ni/Ce-doped Al2O3催化氨分解的转化率约为45.5%,未经Ce 掺杂的Ni/γ-Al2O3的转化率仅为28%。
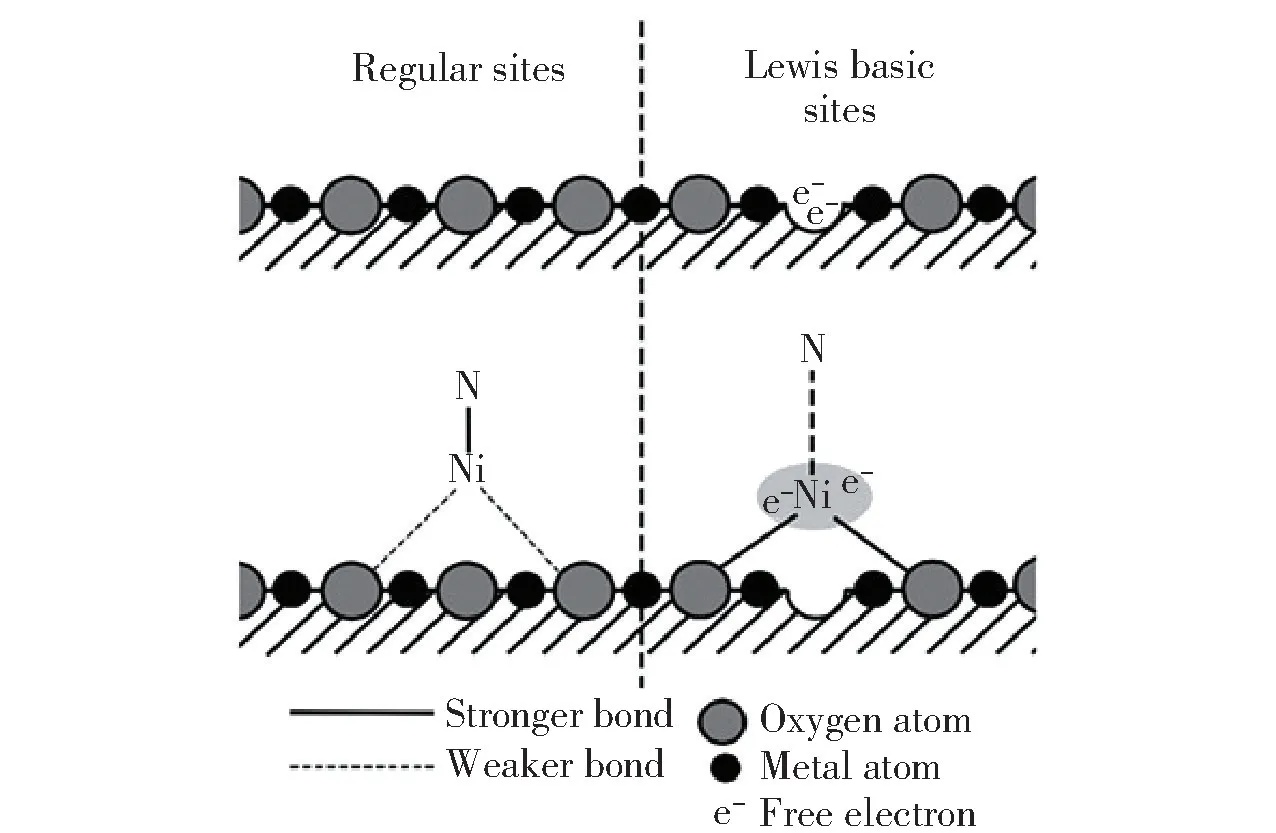
图13 Lewis 碱性位点示意图[104]Fig.13 Schematic diagram of Lewis basic sites[104]
Al2O3作为一种常见载体,其较大的比表面积可以提高金属的分散度、减小粒径尺寸,从而提高催化剂的活性。 通过采用Zr、La、Ce 等元素对其改性则可以提高Al2O3的碱性等性质,从而进一步提高催化剂的活性。
3.3 Mg 的混合氧化物载体
Mg 的混合氧化物是一种碱性载体,对负载金属的分散性较好,因此在氨分解催化剂中也有较多的应用。 Qiu 等[72]采用机械混合法(MM)、溶胶凝胶法(SG)和双层氢氧化物(LDH)法制备了3 种MgAl2O4载体,并对其负载的Ni 催化剂的氨分解性能进行了研究,结果表明,Ni/MgAl2O4-LDH 显示出最高的氨分解性能,在600 ℃、30 000 mL/(g·h)的氨气空速下,NH3转化率可以达到88.7%,H2生成速率达到29.7 mmol/(g·min),MgAl2O4-LDH 对Ni的高度分散作用是该催化剂具有较高活性的原因。Su 等[105]以NiMgAl-LDHs 为前驱体制备了Ni、Mg、Al 物质的量比不同的Nix(MgyAlzOn)催化剂,实验结果表明,随着Ni 和Mg 物质的量比的增加,催化剂的活性先增加后减小,而Mg 和Al 的物质的量比对催化剂活性的影响规律不明显。 Ni0.6(Mg0.29Al0.57On)的活性最高,在500 ℃、30 000 mL/(g·h)的氨气空速下,NH3转化率达到99.3%,H2生成速率达到33.3 mmol/(g·min)。 原因是金属氧化物增大了Ni的分散度,防止其烧结和团聚,提高了催化剂的活性和稳定性;而Ni-Mg 之间的协同作用进一步提高了催化剂的活性,特别是表面Ni 的氢溢流效应增强了Ni 位点上的反应循环。 Podila 等[88]制备了3 种Mg的混合氧化物载体MgX(X =Al、Ce、La),并对其负载的Co 催化剂的氨分解性能进行了研究,结果表明,在Co 负载量为5%、Mg 与X 的物质的量比为2时,3 种载体负载的Co 的活性大小顺序为:MgLa >MgCe >MgAl。 对于MgLa 氧化物体系,在Co 负载量为5%、Mg 与La 的物质的量比为5 时,催化剂的活性最高,在500 ℃、NH3空速为6 000 mL/(g·h)时,NH3转化率约为50%, H2生成速率约为3.34 mmol/(g·min),MgLa 体系较高的碱性是其具有较高活性的原因。 当Mg 与La 的物质的量比为5时,Mg 与La 的相互作用最强,催化剂的比表面积和Co 的分散度最大。
以上研究表明,Mg 的混合氧化物载体较高的碱性和对负载金属的高分散能力可以提高铁系元素氨分解催化剂的活性。 采用双层氢氧化物制备Mg 的混合氧化物载体是提高负载金属分散度的有效方法,通过调控催化剂中金属元素的种类与比例可以进一步提高催化剂的活性。
3.4 SiO2载体
SiO2载体性质稳定,与活性组分的亲和力弱,其特定构型(核壳型结构)和孔隙结构会对催化剂的活性产生影响。
Li 等[77]对微孔SiO2(micro-SiO2)和介孔SiO2(meso-SiO2)包裹的Fe 纳米颗粒(nano-Fe)的催化活性进行了研究,结果表明,在400 ~700 ℃的范围内,催化剂的活性大小顺序为:nano-Fe@ meso-SiO2>nano-Fe@ micro-SiO2>nano-Fe。 3 种催化剂中,介孔SiO2包裹的Fe 纳米颗粒表现出了最高的活性,在600 ℃、30 000 mL/(g·h)的氨气空速下,nano-Fe@meso-SiO2催化氨分解的转化率为85.9%,产氢速率为28.76 mmol/(g·min);在73 h 内,包裹在SiO2中的具有核壳结构的Fe 纳米催化剂比裸露的Fe 纳米颗粒具有更高的稳定性。 原因是SiO2载体防止了Fe 纳米粒子的团聚,而介孔SiO2载体具有更大的孔径,更利于Fe 核的暴露以及反应物和产物的扩散,因而表现出更高的活性。 Yao 等[76]在研究包裹于SiO2中的Ni 纳米颗粒(nano-Ni)时发现,具有核壳结构的Ni@SiO2与Ni 纳米颗粒相比,活性有很大的提高。 在550 ℃、30 000 mL/(g·h)的NH3空速下,Si 与Ni 的原子数比为0.2 的nano-Ni@ SiO2催化氨分解的转化率达到61.8%,产氢速率达到20.7 mmol/(g·min);而在相同条件下Ni 纳米颗粒催化氨分解的转化率仅为20.3%,产氢速率仅为6.8 mmol/(g·min)。 原因是核壳结构形成了微囊反应器,促进了氨的分解。
Li 等[75]对具有核壳结构的Ni@SiO2催化剂的研究结果表明,随着SiO2壳层厚度的增加,催化剂的活性略有下降;增加硅壳的孔隙度,可以提高催化剂的活性。 壳层孔隙结构更发达的SP-1 催化剂(孔体积为0.18 cm3/g) 比SP-0 催化剂(孔体积为0.13 cm3/g)表现出更高的活性:在600 ℃、30 000 mL/(g·h)的氨气空速下,SP-1 催化NH3分解的转化率为78.9%,H2生成速率为26.4 mmol/(g·min);而SP-0 催化NH3分解的转化率为66.3%,H2生成速率为22.2 mmol/(g·min)。
以上研究表明,具有核壳结构的铁系元素催化剂的活性和稳定性都得到了提高,高孔隙度、低壳层厚度是提高核壳型催化剂活性的有利因素,制备具有特定构型(核壳型结构)和孔隙度的载体是提高催化剂活性和稳定性的重要方法。
3.5 分子筛载体
分子筛因具有比表面积大、稳定性高、酸碱性可调等优点,被认为是一种十分有前景的催化剂载体[82]。 Hu 等[82]以ZSM-5 分子筛为载体,研究了SiO2和Al2O3的比例对Fe/ZSM-5 催化剂活性的影响,结果表明,随着SiO2和Al2O3比例的降低,金属与载体的相互作用增强,Fe 在载体上的分散度和Fe的还原势垒提高,催化剂的活性也得到提高。 原因是较低的SiO2、Al2O3比例提高了催化剂的酸性,有利于提高活性位点附近NH3的浓度,从而提高催化剂的活性。 在650 ℃、30 000 mL/(g·h)的氨气空速下,负载量为5%的Fe/ZSM-5 催化氨分解的转化率达到98.7%,氢气生成速率达到33 mmol/(g·min)。值得注意的是,在金属氧化物负载铁系元素的研究[88-89]中,载体的碱性通常被认为有利于提高催化剂的活性,这与Hu[82]等的观点不同。
Li 等[106]对气相SiO2、MCM-41 分子筛和SBA-15 分子筛负载的镍催化剂的研究结果表明,分子筛负载的Ni 比普通硅材质负载的Ni 具有更高的活性。 MCM-41 载体的孔结构限制了Ni 的团聚和烧结,增大了Ni 的分散度,因此表现出比SBA-15 更高的催化活性。 在550 ℃、30 000 mL/(g·h)的氨气空速下,采用离子交换法制备的Ni/MCM-41 催化NH3分解的转化率可以达到47.6%,H2生成速率达到15.9 mmol/(g·min)。 Hu 等[81]采用改性固态离子交换法(MSSIE)制备的Ni/ZSM-5 催化剂也取得了较好的效果。 该方法先用HNO3脱除ZSM-5 中的铝,然后再将脱铝样品与硝酸镍进行机械混合,最后经焙烧制得所需催化剂(图14)。 活性测试结果表明,该方法制备的催化剂具有很高的活性,在650 ℃时,NH3的转化率可以达到97.6%,产氢速率达到32.7 mmol/(g·min)。 原因是在催化剂的制备过程中会有一部分Al 被脱除,脱除Al 后的缺陷使Ni 嵌入ZSM-5 的骨架中,这限制了Ni 的长大,同时也提高了Ni 的分散度,增强了Ni 与载体的相互作用。
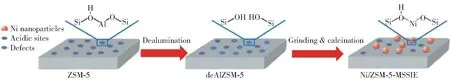
图14 MSSIE 方法制备Ni/ZSM-5 的示意图[81]Fig.14 The illustration of the preparation procedure for Ni/ZSM-5 by the MSSIE method[81]
上述研究表明,分子筛是一种有效的氨分解催化剂载体,通过改变分子筛中的硅铝比例可以实现对催化剂性能的调控,分子筛对负载金属的高分散能力是其提高金属催化活性的重要原因。
4 助剂
铁系元素氨分解催化剂的助剂一般为碱性组分,这些组分包括K、Cs 等碱金属,Mg、Ca 等碱土金属以及稀土元素。 表3 为铁系元素氨分解催化剂的助剂及催化性能比较。
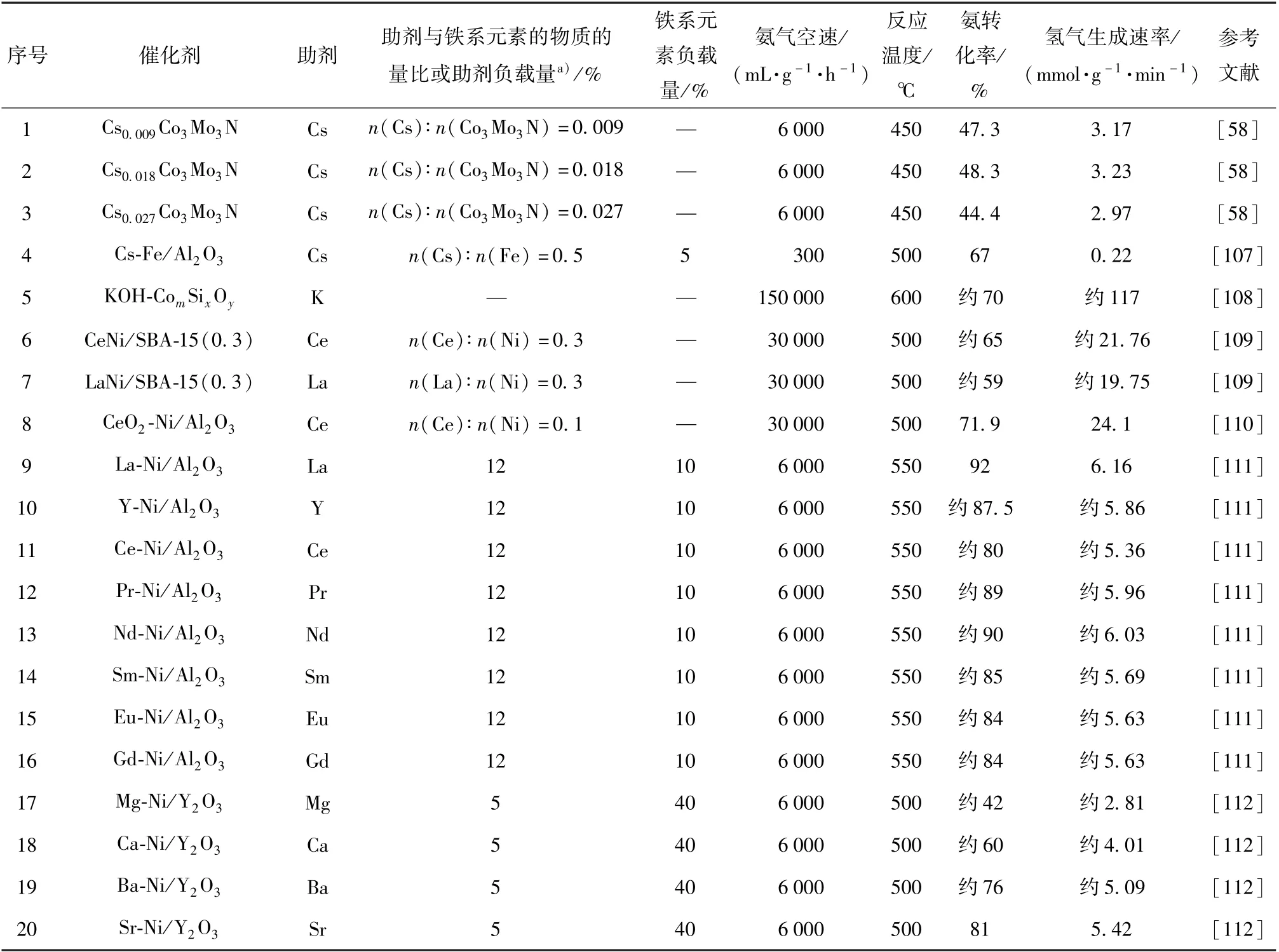
表3 铁系元素氨分解催化剂的助剂及催化性能比较Table 3 Additives and catalytic performance comparison of iron group element-based catalysts for ammonia decomposition
碱金属和碱土金属作为电子供体,可以提高活性组分的电子密度,促进表面N 的脱附,从而提高铁系元素的活性[111]。 Parker 等[107]在研究Cs 对Fe/Al2O3的促进作用时发现,Cs 和Fe 之间存在较强的相互作用,Fe 催化氨分解导致的氢溢流促进了Cs 的还原,而还原性Cs 向Fe 提供电子,弱化了Fe表面N 物种的吸附,促进了表面N 物种的重组脱附。 当n(Cs)∶n(Fe) =0.5 或1 时,Cs 的促进作用最强,催化剂的活性最高。 当Cs 浓度较高时则会形成CsOH,阻塞活性位点,降低催化剂的活性(图15)。 Okura 等[112]对MO-Ni/Y2O3(M =Mg,Ca,Sr,Ba)的研究表明,Sr 和Ba 的加入显著提高了氨分解的活性,但Mg 和Ca 的促进作用不佳,原因是Sr 和Ba 可以和Ni 生成复合氧化物,这种强烈的相互作用有利于电子从Sr 和Ba 向Ni 转移,促进了Ni 表面N 的脱附,从而提高了催化剂活性,而Mg 和Ca与Ni 的相互作用较弱,二者分别以MgO 和CaO 的形式存在。
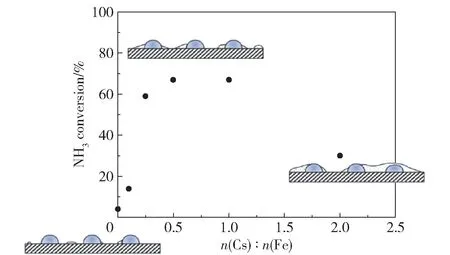
图15 Cs 对Fe/Al2O3的促进作用[107]Fig.15 Promoting effect of Cs on Fe/Al2O3[107]
稀土元素对铁系元素催化剂也有较好的促进效果,但其促进机理与碱金属和碱土金属不同。 Okura等[111]对稀土元素的促进作用进行了研究,结果表明,所有的稀土元素均对Ni/Al2O3有促进作用。 原因是稀土元素可以促进催化剂表面H 的脱附,从而减弱氢的抑制作用,促进氨的分解,其中La 的促进效果最好,原因是其促进H 脱附的能力最强。
除了以上金属助剂外,Li2NH 对铁系元素也有良好的促进作用[113],Li2NH 具有与传统助剂不同的促进机理。 Guo 等[114]的研究表明,Li2NH 的加入使Fe 的活性得到了显著提高(图16)。 Fe 催化氨分解过程中形成的FeNx被认为是Fe 催化氨分解速率较低的原因[12],Li2NH 对Li 存在正向诱导效应,使Li2NH可以与Fe2N 生成三元氮化物Li3FeN2,进而参与反应循环(式(11)和(12)),降低了氨分解的温度。 Bramwell 等[115]对LiNH2(LiNH2可分解为Li2NH)的研究结果表明,Li2NH 对Ni 也有促进作用,在400 ℃时,Ni/C 催化氨分解的转化率仅为19%,而LiNH2-Ni/C 催化氨分解的转化率为53%,转化率提高的原因是Li2NH 与Ni 形成了三元氮化物,促进了氨的分解。
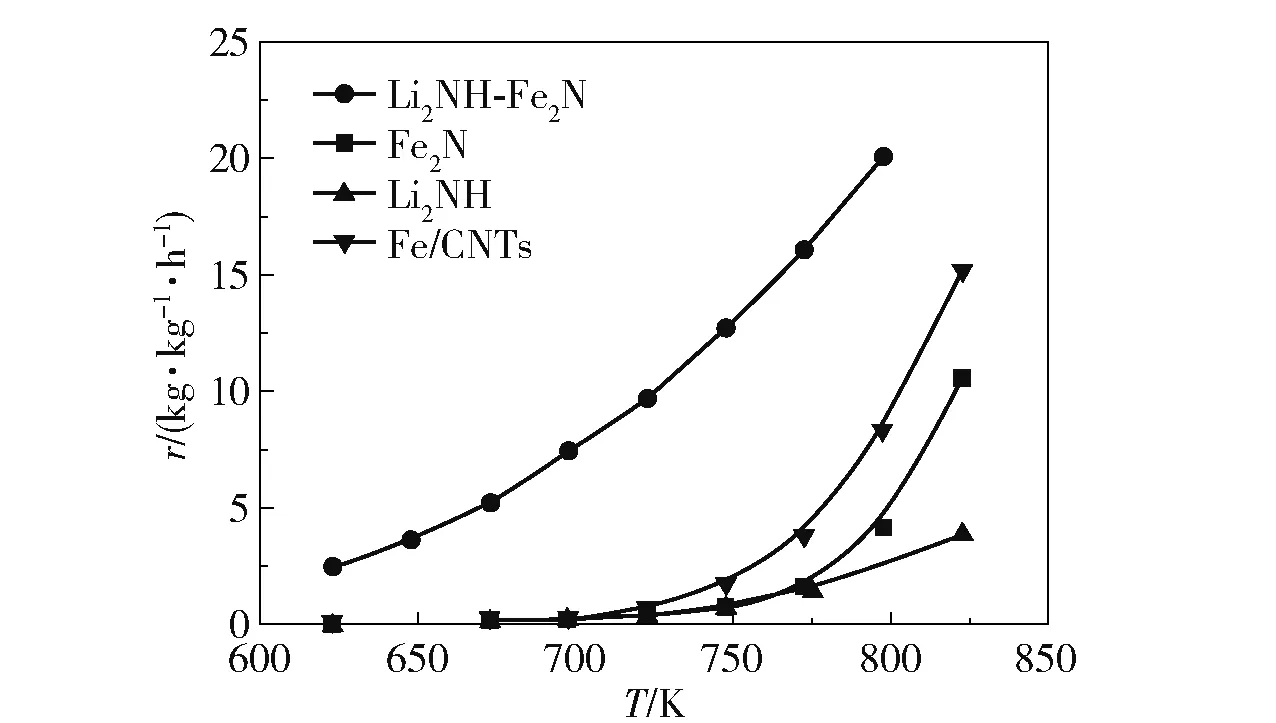
图16 Li2NH 和Fe 基样品催化氨分解的速率[114]Fig.16 Rates of ammonia decomposition catalyzed by Li2NH and Fe-based samples[114]
以上研究表明,碱性助剂在不同催化体系中的最佳种类和比例不同,不同种类的碱性助剂发挥促进作用的机理也不同,因此在选择助剂时,应针对不同催化体系的情况进行具体分析。 Li2NH 具有与传统碱性助剂不同的催化机理,是一种十分有前景的铁系元素氨分解催化剂的助剂。
5 结束语
在全球变暖的大趋势下,氨作为储氢材料日益引起人们的关注。 开发低温高效的氨分解制氢催化剂是实现氨储氢大规模应用的关键一环,本文从氨分解基本原理、催化剂的活性组分、载体和助剂4 个方面对铁系元素氨分解催化剂的研究进展进行了介绍,主要总结为以下几点。
(1)铁系元素催化氨分解是一个逐步脱氢的过程,但氨分解过程的限速步骤仍存在争议(氨脱氢或N 的重组脱附),因此对限速步骤的研究,应根据具体的催化体系进行具体分析。
(2)铁系元素催化氨分解是一个结构敏感反应,催化剂的晶粒尺寸和晶面都会影响催化剂的活性。 晶粒过大或过小都会降低活性,并且在不同的催化体系中,最佳的晶粒尺寸也不同。 对晶粒尺寸的优化需要与活性位点的研究相结合,以尽可能提高活性位点的数量。 不同晶面上原子排列方式的不同导致氨分解中间产物的吸附能不同,进而导致氨分解不同步骤的活化能的差异,最终影响反应速率。对晶粒形貌进行调控可以使催化剂表面暴露不同的晶面种类和比例,因此是提高铁系元素氨分解催化剂活性的重要手段。
(3)铁系元素双金属催化剂的研究取得了一定的进展,单层双金属催化剂的研究为催化剂的开发提供了理论性的解决方案,而铁系元素与贵金属组成的双金属合金则显示出较高的活性。 对铁系元素氨分解催化剂而言,降低氨分解的温度是开发催化剂的重要目标,而双金属催化剂在降低反应温度方面显示出有效性。 例如Fe-Co 双金属催化剂可以将氨完全转化的温度降低至350 ℃,而Ru-Fe 双金属催化剂可以仅在热催化条件下,将氨完全转化的温度降低至500 ℃,表现出与Ru 相似的高活性。
(4)铁系元素氨分解催化剂应用的载体较多,不同载体发挥作用的方式也不同,根据载体的特性选择合适的载体是提高催化剂活性的重要手段。
(5)碱性金属助剂在目前铁系元素催化剂中有较多的应用,不同种类碱性金属的促进机理也不相同。 碱金属和碱土金属可以改善铁系元素的电子结构,而稀土元素可以减弱氢的抑制作用。 近年来的研究发现,Li2NH 与传统助剂的促进机理不同,其促进作用显著,因此值得进一步研究。
根据对以上研究进展的分析,今后对铁系元素氨分解催化剂的研究可以从以下几个方面展开。
(1)在铁系元素催化氨分解的过程中,活性位点对提高催化剂的活性具有重要意义。 高指数晶面具有配位饱和度低的优势,更有利于反应物种的吸附和活化,进而促进氨的吸附和分解。 因此,对高指数晶面的制备与调控可能是提高铁系元素氨分解催化剂活性的一个重要研究方向。
(2)催化剂成本是开发铁系元素氨分解催化剂时一个需要着重考虑的因素,双金属催化剂为铁系元素氨分解催化剂的低成本开发提供了解决方案,特别是开发铁系元素与Ru 等贵金属组成的双金属合金催化剂,这样可以在保持催化剂较高活性的同时,也具有较低的成本,因此是一个具有前景的研究方向。
(3)目前,对铁系元素氨分解催化剂的载体和助剂的研究较多,但载体、助剂和铁系元素的具体作用机理仍不明晰,今后应在持续探索优化不同种类活性组分、载体和助剂的组合的基础上,对各组分进行精确表征和调控,以期降低反应温度,提高催化氨分解的转化率。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
中成药(2019年12期)2020-01-04
石油化工建设(2018年2期)2018-07-11
中国调味品(2017年2期)2017-03-20
凿岩机械气动工具(2016年3期)2016-03-01
上海金属(2015年1期)2015-11-28
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
中学化学(2015年2期)2015-06-05
