
气态碘在COF-103上吸附的理论研究
2024-01-18 02:52童大银赵耀林王禹齐韩子彤喻晨曦
高等学校化学学报 2024年1期
童大银, 赵耀林, 王禹齐, 韩子彤, 王 杰, 张 俊, 喻晨曦
(1. 西安交通大学核科学与技术学院, 西安 710049;2. 大阪大学工学研究科物理学系専攻, 大阪 5650871)
核能作为一种高效、 清洁、 经济的能源, 是世界能源结构的重要组成部分, 核能发电目前已占世界总发电量的9.8%[1]. 随着核电站的大量运行, 其产生的乏燃料累积量不断增加. 在乏燃料后处理首端操作(切割、 溶解)中, 会有大量气态放射碘释放, 其具有易扩散性、 生物毒性和化学毒性[2~4]. 因此, 气态放射性碘的高效捕集对公众健康和环境保护等具有重要意义. 其捕集方法主要有湿法捕集和固体吸附法. 相比于湿法捕集, 固体吸附法操作简单、 对设备耐酸碱腐蚀要求低, 同时放射性废液产生量少[5]. 因此, 固体吸附法被广泛用于世界各国的乏燃料后处理厂. 固体吸附剂在吸附法中发挥着关键作用, 气态碘吸附剂材料主要有沸石类、 活性炭类、 氧化物类、 气凝胶类、 金属有机骨架(Metal organic frameworks, MOFs)和多孔有机聚合物(Porous organic polymers, POPs)材料等[6]. 共价有机框架材料(Covalent organic frameworks, COFs)是多孔有机聚合物材料的一个子类, 具有孔隙发达、 比表面积大、密度低、 热稳定性和化学稳定性高等优点[7], 是最有应用前景的气态碘吸附剂材料之一[4,8,9].
近年来, 气态碘在COFs 材料上吸附性能的研究主要集中在实验方面[10~17]. 如Jiang 等[11]合成了一系列不同孔径大小、 不同拓扑结构且具有一维孔道的二维(2D)共价有机框架材料(包括TPB-DMTP,TTA-TTB, ETTA-TPA和TTA-TFB等). 通过碘分子静态吸附实验, 发现孔体积是决定二维COFs材料碘吸附容量的一个关键因素, 孔体积越大碘分子吸附容量就越大. Zhao等[12]通过溶剂热方法合成了一种具有球形孔腔和多孔壳层的共价有机框架材料SIOC-COF-7, 其对气相和液相中的碘分子均具有较高的吸附容量(4.81和1.27 g/g). 另外, Zeng等[13]也通过溶剂热法合成了两种二维COFs材料(TJNU-201和TJNU-202), 其孔道壁上暴露着苯环和亚胺连接基作用位点, 使得碘分子容易与其发生化学相互作用,因而, 具有超高的碘分子饱和吸附容量(5.625和4.820 g/g). 对气态碘在COFs材料上吸附的理论研究方面, 目前, 相关的文献研究较少[18,19]. 如Lan等[18]通过巨正则蒙特卡罗模拟方法, 计算了187种COFs材料对气态碘的吸附容量, 其中, COF-103材料不仅对甲基碘具有最高的吸附容量(2.8 g/g), 对碘分子的吸附容量也很大(4.98 g/g). 研究结果还表明, 三维(3D) COFs材料对气态碘的吸附性能要优于二维COFs材料, 并且总体上碘分子的吸附容量随COFs孔隙率的增大而增大.
在乏燃料后处理或反应堆严重事故工况下, 可能存在多种杂质气体, 包括气态氧化物(CO, NO,H2O)、 挥发性有机化合物(Volatile organic compounds, VOCs)和氯化物(Cl2和CH3Cl)等[20,21], 其中,VOCs和氯化物主要来源于反应堆严重事故下安全壳内涂料、 线缆或油脂等[22,23]. 在杂质气体存在情况下, 吸附剂对气态碘的吸附性能优劣是评估其实际应用价值的一个关键指标. 对此, 曾有文献报道杂质气体对沸石以及沸石咪唑酯框架材料(Zeolitic imidazolate frameworks, ZIFs)吸附捕集气态碘的影响[20,21,24,25], 但是对COFs材料的影响却鲜有报道.
本文以气态碘分子在COF-103材料上的吸附为研究对象, 通过第一性原理和巨正则蒙特卡罗模拟方法, 研究了碘分子与COF-103的相互作用机理, 并分析了C6H6, CH3Cl和NO等10种杂质气体对碘分子吸附性能的影响.
1 计算方法和模型
1.1 第一性原理计算
1.1.1 计算参数 第一性原理计算采用基于密度泛函理论(Density functional theory, DFT)的维也纳从头算模拟仿真包VASP(Viennaab-initiosimulation package, VASP)进行[26~29]. 原子离子实和价电子之间的相互作用由投影缀加平面波PAW(Projector augmented wave, PAW)方法描述[30,31], 碳、 氢、 硼、 氧、 硅和碘元素的价电子排布分别为2s22p2, 1s1, 2s22p1, 2s22p4, 3s23p2和5s25p5. 电子的交换关联作用由广义梯度近似(Generalized gradient approximation, GGA)中的PBE(Perdew-Burke-Ernzerhof)泛函描述[32], 截断能设置为600 eV. 由于COF-103 晶胞结构实空间较大, 布里渊区K点采样为单个Gamma 点[33~35]. 在结构弛豫过程中, 自洽迭代能量和作用在原子上的力的收敛标准分别为10-6eV和0.2 eV/nm. 此外, 计算中采用DFT-D3的范德华校正方法[36], 通过色散能修正项对体系总能进行修正, 使DFT 计算可描述体系中的范德华相互作用. 体系吸附能(Eads, eV)及长程色散相互作用能(ΔEdisp, eV)采用如下公式计算[20]:
式中:ECOF/I2和(eV)分别为I2吸附后吸附体系的总能和色散能修正项;ECOF,EI2,和(eV)分别为结构弛豫后COF-103和I2的总能以及相应的色散能修正项.
采用差分电荷密度(Δρ, e/Bohr3)和Bader电荷差值(ΔQ, e)对I2与COF-103之间的电荷转移进行了研究, 其计算公式如下:
式中:ρI2/COF,ρCOF和ρI2(e/Bohr3)(1 Bohr=0.05291772 nm)分别为吸附体系电荷密度以及COF-103和I2的电荷密度;Qads(X)和Qvac(X)(e)分别为稳定吸附体系和真空条件下X原子的Bader电荷.
1.1.2 COF-103模型和吸附位点 COF-103初始结构来源于CoRE COFs 结构数据库[37], 其晶胞结构弛豫后的三维模型(a=b=c=2.839 nm,α=β=γ=90°)如图1(A)所示, 是由苯环、 硼-氧六元环(B3O3ring)和SiC4四面体团簇组成, 相应的有机结构单元如图1(B)所示. 根据有机单元结构以及不同气态碘分子I2的吸附取向, 确定的吸附位点包括VO, VB, VC1, VC2, PBC, PBO, PCC和S. 其中, VO, VB, VC1和VC2分别表示I2在O 原子、 B 原子、 C1 原子和C2 原子上垂直吸附的吸附位点, PBC, PBO和PCC分别表示I2在B-C 原子、 B3O3六元环和苯环上平行吸附的吸附位点, S则表示靠近Si原子的吸附位点.
1.2 巨正则蒙特卡罗模拟
巨正则蒙特卡罗(Grand Canonical Monte Carlo, GCMC)计算采用开源分子模拟软件RASPA 2.0 进行[38]. 每个模拟过程包括2×105个平衡循环和2×105个取样循环, 在每次循环中都至少包括20次对吸附质分子随机的平动、 转动、 插入、 删除、 部分再生和完全再生等蒙特卡罗操作, 即每个模拟过程至少包括8×106次蒙特卡罗操作. 在模拟过程中, 吸附质与吸附剂(COF-103)以及吸附质与吸附质之间的非键相互作用采用范德华相互作用和静电相互作用描述. 范德华相互作用由Lennard-Jones(L-J)势函数描述, 截断半径为1.4 nm. 静电相互作用由Ewald 方法描述[39], 计算了包括实空间和倒易空间的静电势能. COF-103模型采用的是DFT弛豫后的晶胞结构, 在x,y,z方向上均采用周期性边界条件. 通用力场(UFF)能够较准确描述气态碘与COFs之间的相互作用[18], 因此, 本文COF-103的力场参数源于UFF力场[40]. I2模型采用联合原子球模型[18,24], 杂质气体分子的模型如图S1(见本文支持信息)所示, 其相应的力场参数如表S1(见本文支持信息)所示. 原子之间的L-J交互作用力场参数采用Lorentz-Berthelot混合规则获得. 另外, COF-103的He孔隙率0.771采用开源软件Zeo++通过探针分子可及可占据孔体积方法(Accessible probe-occupiable pore volume, Ac-PO)计算[41,42], 与文献值0.797 相符[37,43]. COF-103的原子电荷基于DDEC(Density derived electrostatic and chemical)方法通过Chargemol 程序计算[44,45]. 为了评估杂质气体对I2吸附的影响, 计算了COF-103对I2/杂质气体的吸附选择性(Si/j), 计算公式如下[24]:
式中: 下标i和j分别表示i和j组分,X和Y(mmol/g)则分别表示对应组分在主体相中的摩尔分数和吸附相中的吸附量.
2 结果与讨论
2.1 稳定吸附构型及吸附能
首先, 基于第一性原理计算方法研究了碘分子在COF-103上的吸附, 考虑了碘分子垂直和平行的吸附取向. 计算得到的不同位点处的稳定吸附构型如图2(A)~(H)所示, 相应的吸附能、 色散相互作用能以及结构参数等列于表1. 从图2可以看出, 在垂直吸附位点VC1, VC2和VO, 碘分子分别与位点对应的C1, C2和O原子相互作用距离较短. 而在VB位点, 碘分子与苯环上碳原子(I—C: 0.315 nm)相互作用距离较短, 而不是硼原子(I—B: 0.335 nm). 从表1可见, 所有位点的吸附能均为负值, 这说明碘分子在COF-103上各个位点的吸附是稳定的, 且吸附过程为放热反应. 同时, 在吸附位VB, S, VC1和VC2的吸附能最大, 分别为-0.69, -0.70, -0.72和-0.74 eV, 并且在这4个稳定位点处I—C原子间的距离最小, 说明碘分子偏向于吸附在COF-103 苯环结构单元上的碳原子位点处. 这与Banerjee 等[46]采用单晶X射线衍射方法确定的碘分子与苯环具有明显相互作用的结果一致. 此外, 对比未吸附的碘分子I—I键长(0.268 nm), 吸附后的I—I键轻微伸长, 特别是在最稳定的吸附位点VC2, I—I键伸长到0.271 nm. 根据键价理论, 可以得出碘分子吸附后I—I键的强度减弱, 这与文献[19,47]报道的碘分子吸附在ZIFs和COF-DL229上之后其I—I键强度减弱相一致.
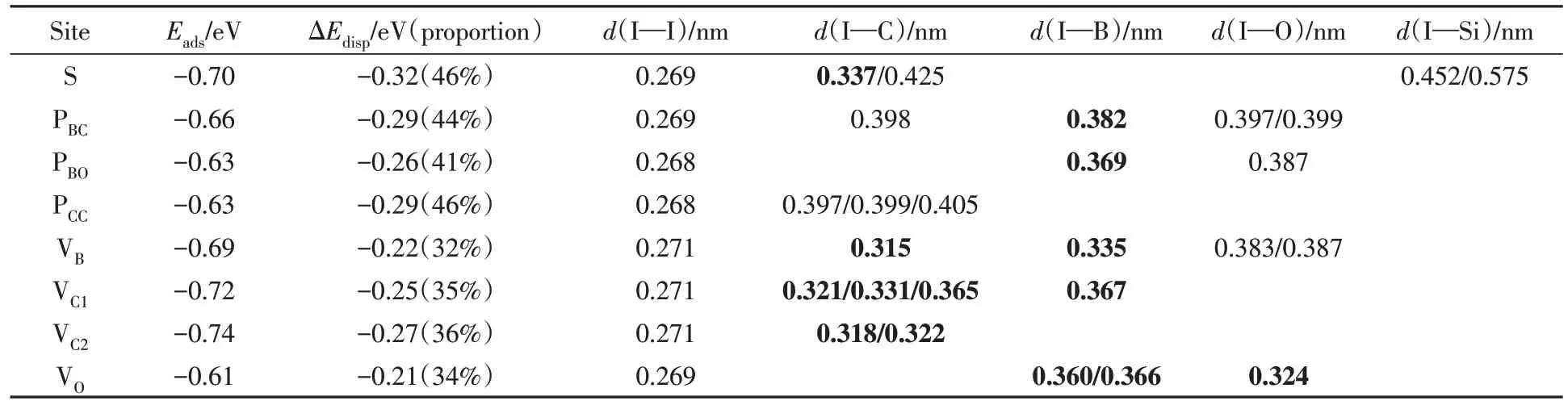
Table 1 Adsorption energies(Eads), dispersion energy(ΔEdisp) and structural parameters(d) of stable adsorption configurations at different sites

Fig.2 Partial structures of stable adsorption configurations at different sites of S(A), PBC(B), PBO(C),PCC(D), VB(E), VC1(F), VC2(G) and VO(H)The unit of structural parameter is nm. I: purple; C: brown; B: green; O: red; H: pink; Si: blue.
从表1还可看出, 吸附能中色散相互作用能的占比较大, 最多可以达到46%, 这说明长程色散相互作用是碘分子与COF-103之间的一个重要相互作用. 此外, 通过对比碘原子与COF-103上碳、 硼、 氧原子的距离及其范德华半径之和(表S2, 见本文支持信息), 可以发现, 碘分子与COF-103之间形成的部分化学键键长介于典型的共价单键键长和范德华半径之和之间[48], 这说明碘分子与COF-103之间形成了次级键[49](在表1中以黑色加粗表示), 即其间也存在次级键相互作用.
2.2 电荷转移和电子结构
为了揭示碘分子与COF-103的相互作用机理, 选取了最为稳定的VB, S, VC1和VC24个吸附位点进行了电荷转移的分析, 包括差分电荷密度及Bader电荷计算[50,51].
4个最稳定位点的差分电荷密度如图3(A)~(D)所示, 黄色部分表示电荷密度增加, 蓝色部分表示电荷密度减小. 可以看出, 吸附后碘分子周围存在明显的黄色区域, 这表明吸附后碘分子周围电荷密度增加, 而COF-103的电荷密度则相对减小. Bader电荷计算结果列于表2. 对比气态碘分子每个碘原子所带的Bader电荷(7.00 e)可以发现, 靠近COF-103的碘原子(I1)电荷有少量减小, 而另一个碘原子(I2)的电荷增大. 总体上, 吸附后碘分子的电荷增加, 而COF-103的电荷减小, 电荷由COF-103转移到碘分子, 这与图3中差分电荷密度的结果相符. 此外, 从表2还可以发现, VC2吸附位点的电荷转移最多(0.07 e), 这与VC2吸附位点的吸附能最大(-0.74 eV)一致.

Table 2 Bader charge results(Q and ΔQ) at favorable adsorption sites

Fig.3 Charge density difference at favorable adsorption sites of S(A), VB(B), VC1(C) and VC2(D)The isosurface value is set as 0.0008 e/Bohr3. I: purple, C: brown, B: green, O: red, H: pink, Si: blue.
在此基础上, 进一步选取吸附能最大的VC2吸附位进行了电子结构的计算. 图4(A)和(B)分别给出了吸附前后碘分子的投影电子态密度(Projected density of states, PDOS), I2分子轨道分布参考我们之前的工作[19,47]. 可以看出, 吸附后I2分子深能级处的σs成键轨道和反键轨道以及费米能级附近的和反键轨道基本没有发生变化, 而σp和πp成键轨道发生了显著变化, 其中,σp的部分轨道电子态向低能级移动到-4.5 eV处, 同时πp的部分轨道电子态向高能级移动到-1.5 eV处, 这说明I2分子的σp和πp成键轨道与COF-103 存在较强的相互作用. 为了进一步分析相互作用机理, 计算了I2分子吸附在VC2位点处的局域电子态密度图(Local density of states, LDOS), 结果如图5 所示, 其中, I1 和I2 分别为距离COF-103 较近和较远的碘原子, C1 和C2为COF-103 上靠近碘分子的两个碳原子,具体的结构如图S2(见本文支持信息)所示. 从图5 可见, 在σp和πp成键轨道劈裂弥散的范围内(-5~-1 eV), 碘原子和碳原子之间有数个态密度峰重叠, 发生了轨道杂化, 这说明I 和C 之间存在弱的共价相互作用[52~54].

Fig.4 Electronic projected density of states of I2 before(A) and after adsorption(B) onto COF-103 at VC2 siteTDOS: the total density of states. The black dash line denotes the Fermi level.
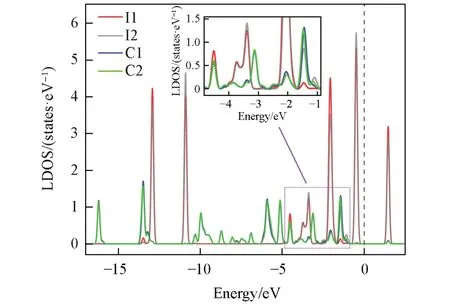
Fig.5 Electronic local density of states of I2 adsorption onto COF-103 at VC2 siteThe black dash line denotes the Fermi level.
2.3 杂质气体影响的DFT计算
采用DFT方法计算了CO, NO, H2O, CH4, C2H6, CH3OH, C2H5OH, C6H6, Cl2和CH3Cl等杂质气体在COF-103 上的吸附能, 通过对比它们与碘分子吸附能的差异, 分析了杂质气体对气态碘吸附的影响.鉴于COF-103的有机结构单元是由苯环和硼-氧六元环组成, 因此, 杂质气体在COF-103上的吸附计算主要考虑了苯环和硼-氧六元环这两个吸附位, 获得的结果列于表3. 对于极性小分子CO, NO和H2O,初始吸附取向为垂直吸附, 并考虑它们与吸附位点不同原子(B, O和C)的相互作用; 而对于非极性分子Cl2, CH4以及具有较复杂空间构型的分子CH3OH, C2H6, C2H5OH, C6H6和CH3Cl, 初始吸附位置于苯环和硼-氧六元环的邻近上方, 接近于平行吸附. 不同吸附取向或相位对杂质气体吸附的影响, 后续将进一步深入研究.

Table 3 Adsorption energies(Eads) of contaminants at phenyl and B3O3 ring site on COF-103
从表3可以看出, 杂质气体中C6H6的吸附能最大, 并且与碘分子在VB, S, VC1和VC2等位点的吸附能大小相近, 这说明COF-103对苯分子的亲和性与碘分子的强弱相当, 即苯分子可能对碘分子在COF-103上的吸附产生较大的影响. 与之相反, COF-103对NO, CO和CH4等的吸附能较小, 与碘分子的吸附能差值较大, 这说明N, CO 和CH4等与COF-103 的亲和性较低, 对碘分子在COF-103 上吸附的影响较小. 需要注意的是, 通过吸附能的计算对比来分析杂质气体对气态碘吸附的影响虽然具有一定的合理性, 但是这种方法是间接的, 只考虑了杂质气体或碘分子单组分吸附的情况, 未考虑杂质气体与碘分子共存的吸附情况, 同时, 也忽略了杂质气体与碘分子可能的协同吸附效应.
2.4 杂质气体影响的GCMC计算
为了更直接、 准确地揭示杂质气体对COF-103吸附气态碘的影响, 采用巨正则蒙特卡罗方法进一步模拟研究了杂质气体与碘分子在COF-103上的双组分吸附过程, 并计算对比了相应的吸附量(Qab)以及吸附选择性. 其中, 模拟温度参考乏燃料后处理厂气态碘过滤装置的运行温度, 设为150 ℃[55]. 考虑到气态碘和杂质气体在乏燃料后处理厂废气中的分压较低, 模拟总压力设置为在当前温度下碘分子的饱和蒸气压40 kPa[56]. 为了准确反映杂质气体、 碘分子和COF-103 之间相互作用强度对结果的影响,通过RASPA程序设置, 气态碘和杂质气体的初始摩尔分数比设为1∶1.
计算得到的COF-103对气态碘分子和杂质气体的绝对吸附量和吸附选择性分别如图6(A)和(B)所示, 具体数据列于表S3(见本文支持信息). 在图6(A)中, 蓝色点线、 红色点线分别表示等摩尔比双组分情况下碘分子和杂质气体的绝对吸附量, 蓝色虚线为相同温度和分压下碘分子单组分吸附的绝对吸附量. 可见, 在杂质气体为苯分子的情况下碘分子的吸附量降低最明显, 而在其它杂质气体存在的情况下碘分子的吸附量与单组分碘分子的吸附量差别不大. 同时, 在所有杂质气体中, 苯的吸附量(6.66 mmol/g)最大, 而其它杂质气体的吸附量均较小, 不超过0.5 mmol/g. 这说明苯分子能够显著影响碘分子在COF-103上的吸附, 而其它杂质气体的影响相对较小. 图6(B)给出了等摩尔比双组分情况下的吸附选择性, 即吸附相中碘分子和杂质气体的摩尔分数比. 通常, 吸附选择性越低, 该杂质气体对碘分子吸附的影响越大. 如图6(B)所示, 苯分子对应的吸附选择性最低, 符合图6(A)显示的对碘分子吸附量有很大影响的结果. 氯气和甲基氯对应的吸附选择性也较低, 同样, 由图6(A)也可见, 这两种气体对碘分子吸附量的影响相比其它杂质气体也稍大. 同时, 结合图6(A)还可看出, 由于CH4, CO和H2O在COF-103上的吸附量很小, 并且其对碘分子的吸附量几乎没有影响, 因此这3种杂质气体对应的吸附选择性均相对较高, 这与文献[57,58]中水分子几乎不影响碘分子在COFs上吸附的实验研究结果一致.

Fig.6 Absolute adsorption loading of I2 and contaminants(A) and adsorption selectivity(B) in case of equimolar binary adsorption at 423.15 K
为了揭示COF-103吸附碘分子过程中苯分子的影响机理, 计算了等摩尔比双组分情况下碘分子和苯分子在COF-103上稳定吸附时的质心密度(Center of mass, COM)分布, 结果如图7所示. 其中, 红色和蓝色部分分别表示较强和较弱的吸附位点, 并且每个质心密度分布图都单独作了归一化处理, 只能反映吸附质的相对密度分布. 可以发现, 图7(A)和(B)中的吸附区域存在明显重叠, 这说明在双组分情况下苯分子会占据碘分子的部分吸附位点, 从而导致碘分子的吸附量降低.

Fig.7 COM distribution of iodine molecule(A) and benzene(B) on COF-103 in the case of equimolar binary adsorption at 423.15 K
为了进一步探究杂质气体对碘分子在COF-103 上吸附的影响机理, 计算了杂质气体的等量吸附热(Qst, kJ/mol)及其与碘分子的等量吸附热差值(Difference in isosteric heat of adsorption, DIH). 为了与等摩尔比双组分吸附的研究结论进行对比分析, 计算温度为150 ℃, 压力为20 kPa.Qst[24,59,60]及DIH(kJ/mol)的计算如下:
式中:U(kJ),N(mol),T(K)和R(0.008314 kJ·mol-1·K-1)分别表示吸附相的势能、 吸附量、 温度和气体常数;表示在巨正则系综下对其求平均;Qs(tI2)和Q(stContaminant)分别表示碘分子和杂质气体的等量吸附热. 等量吸附热表示单位数量吸附质分子吸附时放出的热量, 一定程度上可以表示吸附质分子和吸附剂材料的亲和性. 图8灰色柱状图为不同杂质气体的等量吸附热. 可见, 杂质气体等量吸附热按 C6H6>Cl2>CH3Cl>CH3OH>C2H5OH>C2H6>NO>CH4>CO>H2O 的顺序依次减小, 具体数据列于表S4(见本文支持信息). 可以发现, 苯分子的Qst(51.80 kJ/mol)最大, 表明苯分子与COF-103 的亲和性很高, 会与碘分子发生明显的竞争吸附, 导致碘分子的吸附量显著下降.相比于碘分子在同等温度和压力下的Qst(59.35 kJ/mol), 其它杂质气体的Qst都较小(9.20 kJ/mol <Qst<17.81 kJ/mol), 说明其与COF-103 的亲和性较低, 不易影响碘分子的吸附. 结合图8 的DIH 和图6(B)的吸附选择性S可见, 总体上碘分子与杂质气体的等量吸附热差值越大, 相应的吸附选择性就越高.

Fig.8 Isosteric heat of adsorption for various contaminants and difference of isosteric heat at 423.15 K
3 结论
通过第一性原理方法和巨正则蒙特卡罗模拟, 对气态碘分子在COF-103 上的吸附行为进行了系统研究, 阐明了气态碘分子与COF-103 的相互作用机理以及杂质气体的影响机理. 稳定吸附构型和吸附能的计算结果表明, 碘分子在COF-103上的吸附过程为放热反应, 并且最稳定的吸附位为VC2. 长程色散相互作用是碘分子与COF-103之间的一个重要相互作用, 其对吸附能的贡献可以达到46%. 碘分子在COF-103上吸附后会形成I—C, I—B或I—O等次级键. 电荷转移和电子结构的计算结果表明,从COF-103到碘分子之间存在少量的电荷转移. 此外, 在最稳定吸附位点VC2, 碘分子的σp和πp成键轨道会发生劈裂, 并与COF-103上碳原子的价电子轨道发生杂化, 表明碘原子和碳原子之间形成了弱的共价相互作用. 杂质气体吸附的DFT和GCMC计算结果表明, 苯分子与COF-103的亲和性最高, 并且苯分子能够占据碘分子的吸附位, 从而显著降低碘分子的吸附量. 而其余杂质气体与COF-103的亲和性较低, 对碘分子吸附的影响不大. 研究结果有助于理解气态碘在COFs 上的吸附机理以及杂质气体的影响, 对高效气态碘吸附剂的筛选、 设计和合成等具有一定的理论指导意义. 此外, 液相中碘的吸附去除也具有重要的工业应用价值, 采用第一性原理方法开展模拟计算, 可进一步拓展COFs材料在相关领域的应用前景.
支持信息见http: //www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230401.
感谢大阪大学工学研究科物理学系専攻Yoshitada Morikawa教授和Azim Fitri Zainul Abidin博士及中广核研究院有限公司陈忠村博士的帮助.
猜你喜欢
中学生数理化·中考版(2021年10期)2021-11-22
艺术品鉴(2020年6期)2020-12-06
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中国特种设备安全(2020年11期)2020-06-09
柴油机设计与制造(2018年3期)2018-10-13
柴油机设计与制造(2018年2期)2018-08-29
柴油机设计与制造(2018年1期)2018-04-20
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
新高考·高一物理(2015年6期)2015-09-28
