
DNA碱基编辑技术的研究进展及在猪基因修饰中的应用
2024-02-05 06:12杨帅朋屈子啸朱向星唐冬生
生物技术通报 2024年1期
杨帅朋 屈子啸 朱向星 唐冬生,
(1. 佛山科学技术学院生命科学与工程学院 广东省动物分子设计与精准育种重点实验室,佛山 528225; 2. 佛山科学技术学院医学院 广东省基因编辑工程技术研究中心,佛山 528225)
基因编辑技术被誉为21世纪生命科学领域最具有突破性和革命性的生物技术之一。它能够对目标染色体序列或目的基因进行定点重组、敲除、插入或替换,从而影响基因的自然表达,以达到研究基因功能的目的[1]。自基因编辑技术诞生以来,就被迅速运用于基因功能探索、动植物遗传改良和疾病治疗,极大推动了生命科学及其相关学科的发展。基因编辑首次报道于1971年,Danna团队成功使用流感嗜血杆菌核酸内切酶(endonuclease)R切割SV40的DNA[2],自此之后,越来越多的工具酶相继被开发并应用。其中应用最广泛的有1996年开发的第1代锌指核酸酶(zinc finger nucleases,ZFN)基因编辑技术[3];2010创建的第2代类转录激活因子效应物核酸酶(transcription activator-like effector nucleases, TALEN)基因编辑技术[4];2012年问世的第3代由成簇的规则间隔短回文重复系列(clustered regularly interspaced short palindromic repeat,CRISPR)及CRISPR相关核酸酶蛋白(CRISPR-associated protein, Cas)组成的新型基因编辑器[5]。三代基因编辑器同时存在负责识别特定序列的识别域和负责切割DNA序列的切割域。在ZFN系统中,由锌指蛋白负责识别、Fok I核酸内切酶负责切割[6];TALEN系统中则是由转录激活因子类效应因子(transcription activator-like effector, TALE)作为识别模块,Fok I酶作为切割模块[7];CRISPR/Cas系统则包含了负责识别的crRNA(CRISPRRNA)和负责切割的效应蛋白内切酶Cas两部分[8]。伴随着技术的发展迭代,科技的进步,以CRISPR/Cas9为代表的第3代基因编辑器因其技术门槛低、操作便利、载体构建简单、编辑效率高和成本低廉等特点受到了广大科学家们的青睐,成为目前使用最为广泛的基因编辑技术[9-10]。
前三代基因编辑工具均是在识别结构特异性基因组DNA序列后,由切合蛋白负责切割靶向基因组DNA,使靶点序列处产生双链断裂(double-stranded break, DSB),诱发生物体内源修复途径,包括非同源末端连接(non-homologous end joining, NHEJ)和同源重组(homologous recombination, HR),从而实现靶点的定点敲除、替换、插入等精准修饰[11-12]。其中NHEJ途径是主要的修复途径,该途径优缺点十分鲜明,修复迅速但错误率高,常常会在靶点附近引入少量碱基的插入或缺失(insertions and deletions, Indels),从而对基因原有功能造成不可逆的破坏。HR具有更高的修复精准性,但是操作复杂且局限性大。主要是因为HR途径需要人为插入与目标靶基因同源的DNA序列,而且该过程主要发生在细胞复制间期的S期和G2期[13]。虽然HR途径可以解决大部分生活生产和研究中遇到的基因修饰问题,但在基因编辑过程中,不能保证仅仅发生HR修复,因此,就会引入不必要的Indels影响基因的正常功能。而且,如何将供体有效的送往细胞体也是另一个亟需解决的难题,因此,基因编辑技术在日常生产研究中受到一定制约[11-12,14]。
然而,大多数人类遗传疾病的发生和动植物性状的改变均是由单核苷酸变异产生的[15-16],且依赖DSB途径的传统基因编辑技术易引入Indels,产生不可预测的基因突变,因此,传统基因编辑器不是疾病治疗或动植物遗传育种资源开发的理想工具。科学家们迫切需要开发出一种新的能够精准高效完成碱基替换的基因编辑系统。2016年,哈佛大学的David Liu团队将理想变成了现实,他们将大鼠(rattus norvegicus)的胞嘧啶脱氨酶(APOBEC1)、具有单链切割活性的nCas9(Cas9 nickase)蛋白和具有抑制DNA修复功能的尿嘧啶糖基化酶抑制子(uracil DNA glycosylase inhibitor, UGI)组合,成功开发出了第一代单碱基编辑器BE3(base editor 3),BE3的问世,正式拉开了单碱基编辑技术的序幕[17]。截至目前,科学家们相继开发出了多种DNA单碱基编辑器,极大地丰富了碱基编辑器家族成员,主要包括能够实现C T碱基和G A碱基变换的胞嘧啶碱基编辑器(cytosine base editor, CBE)[17];能够转换A T碱基为G C碱基的腺嘌呤碱基编辑器(adenine base editor, ABE)[18-19];C G或C A碱基之间颠换的糖基化酶碱基编辑器(glycosylase base editor, GBE)[20];能够实现A到C或T颠换的腺嘌呤碱基颠换编辑器(adenine transversion base editor, AYBE)[21];能够同时实现C T碱基、G A碱基突变和A G碱基、T C碱基突变的双碱基编辑器(dual base editor,DBE)[22-25];能够实现所有碱基间相互转换的引导编辑器(prime editor, PE)[26](图1)。
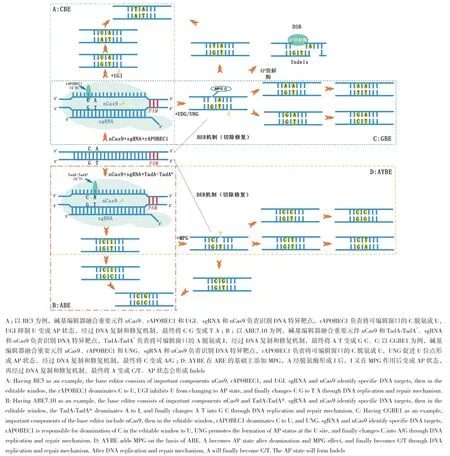
图1 碱基编辑器工作原理示意图Fig. 1 Mechanisms of base editor systems
1 碱基编辑器的特点
碱基编辑器的核心元件是由已经失去切割活性的或只有单链切割活性的Cas蛋白和作用于单链DNA(single-stranded DNA,ssDNA)的工具酶组成。该系统由sgRNA(guide RNA)引导定位,在不引入DSB、无需供体DNA参与的情况下,通过工具酶的酶促反应,借助生物体内DNA光修复和复制机制,从而实现对目标靶点的精确修饰。与传统基因编辑相比,碱基编辑技术有以下优点:(1)能够有效避免引入Indels,从而减少细胞毒性,减少细胞紊乱发生几率。科研工作者们也不需要花费过量的时间和精力寻找解决Indels的危害。(2)无需供体DNA参与。因此,节约了大量设计供体DNA的时间和减少了投递供体DNA的难度。(3)技术门槛低。碱基编辑器同时兼具安全高效和普适性的特点,因此自进入大众视野以来便被迅速而广泛地应用于各领域[27](表1)。也因此,碱基编辑技术入选Science杂志2017年度十大科学突破之一(http://vis.sciencemag.org/breakthrough2017/)。
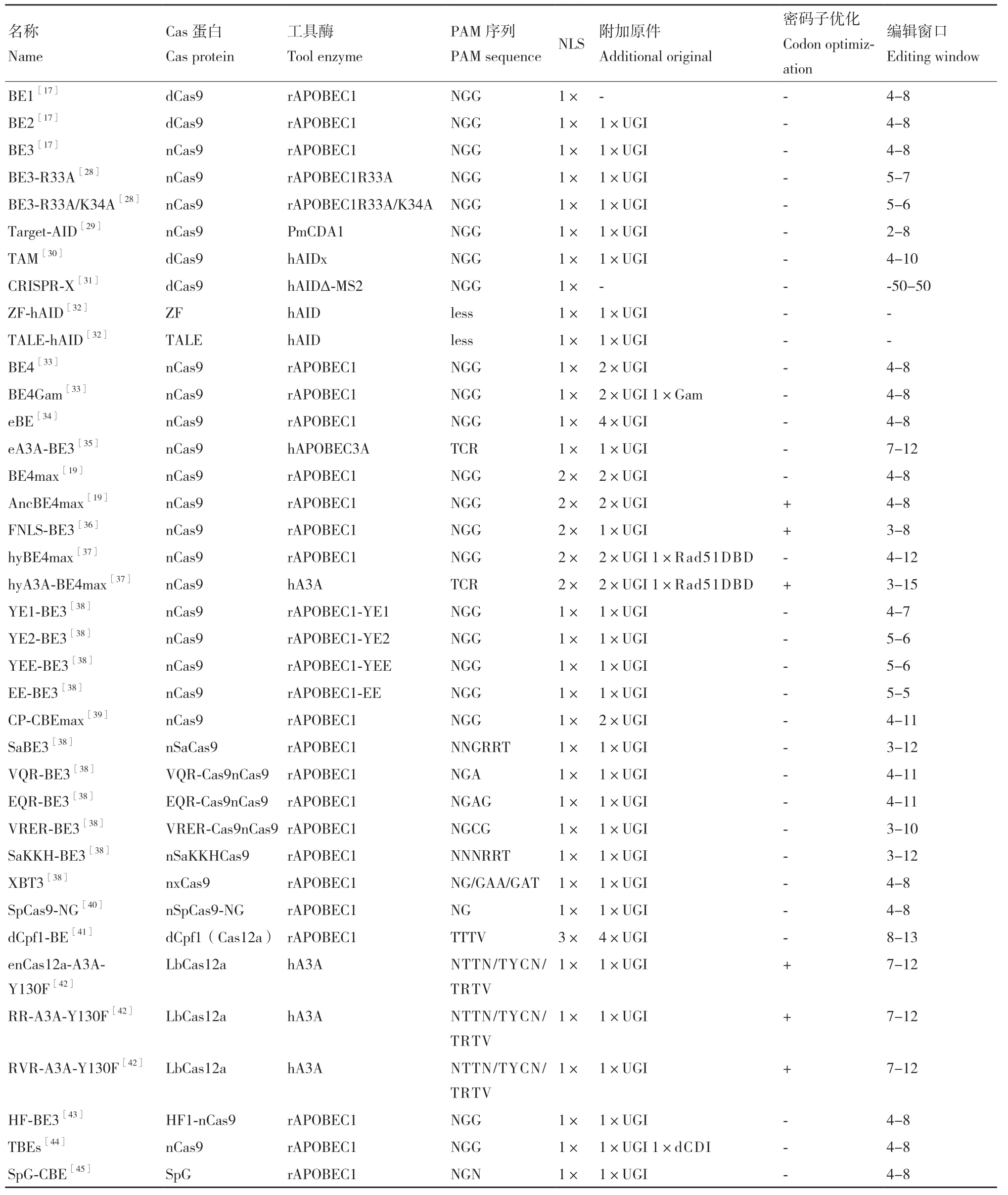
表1 各种碱基编辑器的特点Table 1 Characteristics of each base editor
2 碱基编辑器系统
2.1 胞嘧啶碱基编辑器CBE
2.1.1 CBE的建立 碱基编辑器起源于CRISPR/Cas9系统,是更加精准和精细的新型基因编辑系统。由于CRISPR/Cas9工作时需要引入Indels,导致基因组较大片段的改变,如易位或删除[55-56]或是激活细胞内p53通路[57]。为了避免Indels的引入,哈佛大学David Liu团队开创性的把改造后的丧失核酸酶功能的Cas9蛋白和仅作用于单链DNA的rAPOBEC1融合,首次构建出了第一代胞嘧啶碱基编辑器BE1,BE1的编辑活性窗口位于PAM(NGG)区域上游第13-17位碱基之间约5 nt的区域[17]。BE1的工作原理是在sgRNA引导下,当编辑器结合到目标靶点时,编辑窗口内的胞嘧啶(C)在rAPOBEC1作用下被水解脱氨形成尿嘧啶(U),在DNA的复制和修复过程中U被认为是胸腺嘧啶(T),在互补链中相应位置的鸟嘌呤(G)会被替换成腺嘌呤(A),再经过一轮复制,改变后的A与T互补配对。结果是原先DNA中的C被T取代,G被A取代,实现了C G碱基对被T A碱基对取代。随着该团队在纠正人类细胞疾病相关突变测试时发现,BE1在细胞中编辑效率仅为0.8%-7.7%,与体外实验相比,效率降低了5-36倍。推测可能的原因是体内尿嘧啶DNA糖基化酶(uracil DNA glycosylase, UDG)进行的切除修复(base-excision repair, BER)造成的,遂该团队把枯草芽孢杆菌噬菌体的尿嘧啶DNA糖基化酶抑制蛋白(uracil DNA glycosylase inhibitor, UGI)嵌入到BE1的3'末端,成功研发出第2代碱基编辑器BE2,在相同位点的测试中表明,BE2的平均编辑效率约为20%,是BE1的3倍。随后的数据还显示,BE1和BE2系统的Indels形成率不到0.1%。为了克服真核生物的错配修复机制(mismatch repair, MMR),提高编辑效率,该团队恢复了BE2中Cas9 蛋白HNH结构域840位置处的催化His残基,从而产生了第3代碱基编辑器BE3。相比于BE2,BE3在人类细胞中的编辑效率提高了2-6倍,约为37% C T(G A)得到转换。
几乎同时,日本神户大学的Nishida团队也开发出了一种类似于BE系统的碱基编辑器,研究人员称之为Target-AID,经测试,Target-AID系统能够在酵母细胞和哺乳动物细胞中实现有效的基因定点替换[29]。Target-AID与BE3有所不同的是,Target-AID则是采用了七鳃鳗的胞嘧啶脱氨酶,而且UGI和脱氨酶(PmCDA1)均连接在dCas9或nCas9的3'末端。且该系统拥有较低的细胞毒性但是编辑窗口略显狭小。
2016年,3篇基于融合人类胞嘧啶脱氨酶hAID的碱基编辑器创立成功的消息先后被发表[30-32]。分别是中国科学院上海生命科学研究院常兴团队开发的TAM系统(Targeted AID-mediated mutagenesis)[30]、美国斯坦福大学Michael C Bassik团队开发的CRISPR-X系统[31]以及美国哈佛大学George M Church团队开发的与基因编辑技术融合的碱基编辑器[32]。TAM系统[30]融合了dCas9及UGI,在sgRNA的指引下就可以完成大范围基因编辑;CRISPR-X系统[31]仅融合了dCas9,并没有融合UGI,CRISPR-X系统是通过MS2招募hAID脱氨酶,通过创建基因组多位点定点突变文库的方式可以同时靶向基因组的多个位点,完成基因组多位点的定点突变,且存在较少的脱靶现象。TAM系统和CRISPR-X系统在结合多组sgRNA的条件下,可在哺乳动物细胞中实现较大范围(编辑范围可以达到100 nt)以及较多类型的碱基替换。George M Church团队开发的碱基编辑器是将前两代基因编辑器和单碱基编辑器融合[32]。将ZFN或TALEN的特异性识别单元与hAID脱氨酶融合,构建了全新的碱基编辑器。与融和了Cas9蛋白变体的碱基编辑器相比,George M Church开发的碱基编辑器则不受PAM序列的限制,但在大肠肝菌及人类细胞测试中发现编辑效率普遍不高(大肠杆菌中13%,人类细胞中2.5%),推测形成这种现象的原因可能是因为ZFN或者TALEN与DNA结合后无法形成ssDNA,仍然保持双链DNA(double-stranded DNA, dsDNA)的形式,无法形成足够数量的可用于hAID脱氨酶催化反应的底物,因此该系统的编辑效率不高。
2.1.2 CBE的优化 CBE系统的优化和改进的目标主要涉及6个方面:(1)提高目标产物纯度,降低阴性产物的浓度;(2)进一步提高CBE的表达效率;(3)扩大或缩小可编辑窗口的范围,提高编辑的精准度;(4)进一步提高编辑可编辑窗口的效率;(5)降低碱基系统的脱靶效应;(6)降低碱基系统对背景序列的依赖性。
2.1.2.1 提高目标产物纯度 众多研究结果表明,CBE系统编辑产物中含有其他杂质,产生的原因主要有:(1)CBE除了将C转换为T之外,有一定几率将C突变为G或A[11]。这是因为在UDG作用下,中间体U被切除,暂时形成无嘧啶位点(apyrimidinic site, AP),在经过DNA修复机制及复制作用后,有一定的几率将该位点引入其他碱基,导致原始C突变为其他碱基;(2)在CBE行使功能过程中,会产生少量的Indels[11,17]。原因是编辑过程中产生的AP位点经AP裂解酶作用或自发断裂形成缺口,而在nCas9蛋白切割时,非编辑链上也存在一个缺口,两个缺口正好可形成一个DSB,经过NHEJ修复途径后即产生了Indels。
通过增加UGI的拷贝数、添加游离的UGI分子或优化胞嘧啶脱氨酶,是降低编辑副产物的有效手段。David Liu团队构建的BE4在BE3系统的Cas9的C端多融合了一份UGI拷贝,大幅度提高了C转换效率(约0.5倍),且副产物的比例显著降低[33]。随后,David Liu团队又在BE4的基础上融合了DSB末端保护蛋白Gam,成功构建了BE4Gam,进一步大幅度降低了Indels发生频率(约2.1倍)[33]。基于同样的策略,Wang等[34]的增强型碱基编辑器(enhanced base editor, eBE)是将BE3系统与游离的UGI分子组合,在有效降低副产物的同时提高了BE3的编辑准确度。Geheke等[35]在人类胞嘧啶脱氨酶hAPOBEC3A的基础上改造出了eA3A-BE3,该系统缩小了可编辑窗口,从而减少了旁观者效应。
2.1.2.2 提高编辑活性 研究发现,碱基编辑器的活力水平是影响编辑效率的重要因素之一。在编辑器中,通过优化碱基编辑器的表达原件或者引入其他功能性原件,是提高编辑活性的有效策略。Koblan等[19]通过在BE4的rAPOBEC1-Cas9的N端和C端添加核定位信号构建和优化密码子序列,先后构建了新系统BE4max和AncBE4max,在测试对先天性1f型糖基化障碍疾病治疗研究时发现,AncBE4max校正效率为BE4的2倍。通过变换NLS位置和数量,结合优化nCas9密码子序列的策略,构建了FNLSBE3系统,与BE3系统相比,在哺乳动物细胞的编辑效率平均提高了15倍[36]。Zhang等[37]在Cas9蛋白和脱氨酶之间嵌入了Rad51非序列特异性单链DNA结合结构域,从而得到了编辑活性显著提高、编辑窗口更宽的hyBE4max和hyA3A-BE4max系统。
2.1.2.3 扩大或缩小编辑窗口 不同类型碱基编辑器有不同的编辑窗口,不同的研究目的需要选择不同的编辑靶点,这是精准医疗和育种所要求的。通过寻找新的胞嘧啶脱氨酶或者改造现有的脱氨酶,获得不同的脱氨酶变体是达到改变编辑窗口的有效策略。Kim等[38]突变了脱氨酶关键位点,构建了CBE系统变体 YE1-BE3、YE2-BE3、YEE-BE3和EE-BE3,成功将BE3的编辑窗口从5 nt缩小至1-2 nt,但对靶点位点的编辑效率有所降低。David Liu团队使用循环置换Cas9变体构建而成的CP-CBEmax将编辑窗口从4-5 nt覆盖到8-9 nt,同时减少了副产物的生成[39]。Nishida的Target-AID系统[29]编辑窗口则为3-5 nt碱基。常兴[30]的TAM系统和Michael C Bassik的CRISPR-X系统[31]在融合多组sgRNA的情况下,在哺乳动物细胞中可以实现高达100 nt以及较多类型的碱基编辑。
2.1.2.4 拓宽PAM序列范围 碱基编辑器多是融合SpCas9蛋白,因此只能识别NGG形式的PAM序列。为此,科学家们需要寻找出新的SpCas9蛋白替代蛋白或同功能变体,通过识别不同特异性序列的蛋白体识别不同PAM序列,从而达到解除CBE系统在应用过程中被PAM序列限制的目的。David Liu实验室用Cas9蛋白的不同变体先后创建出了能够识别不同PAM序列的编辑器[38]。分别为NNGRRT形式的SaBE3、NGA形式的SpVQR-Cas9、NGAG形式的SpEQR-Cas9、NGCG形式的SpVRER-Cas9、NNNRRT形式的SaKKH-Cas9以及可以同时识别多种PAM序列形式(NG、GAA和GAT)的xCas9,相比于只能识别NGG序列的SpCas9,xCas9的应用范围更加广泛[58]。Nishimasu等[40]也构建出了能够识别NG形式的SpCas9-NG。Li等[41]则是构建了特异性识别PAM序列TTTV的dCpf1-BE(Cas12a),并且dCpf1-BE可以在pCas9-BE不能编辑的AT富集(AT-rich)区域编辑,填补了spCas9-BE仅能在GC富集(GC-rich)区域实现编辑的不足。赖良学团队将LbCas12a与高活性的脱氨酶结合,构建出了可以识别具有NTTN、TYCN和TRTN形式PAM的编辑系统(enCas12a-、RR-和RVR-A3A-Y130F)[42]。
2.1.2.5 降低碱基系统的脱靶效应 2019年,CBE系统在全基因组中的脱靶问题相继被报道[59-60],其安全问题进入了大众的视野。但是在此之前,科学家们就已经陆续发现CBE系统存在轻微的脱靶现象,并对此展开了研究[61-62]。2017年,David Liu实验室将Cas9蛋白的突变体HF1-Cas9与BE3融合,成功地构建出高保真型BE3系统(High-fidelity BE3,HF-BE3),HF-BE3可明显降低BE3系统对非靶标位点的活性,提高序列编辑的特异性[43]。Lee等[63]在大肠杆菌埃希菌株中通过改造和筛选,成功筛选出了具有高特异性的Sniper-Cas9蛋白,有效地降低了脱靶现象。Wang等[44]结合胞嘧啶脱氨酶的抑制序列结构域(deoxycytidine deaminase inhibitor,dCDI)开发的碱基编辑器tBEs可以对非靶标位点进行锁定,仅仅在靶点位置保持开放,从而降低了Cas9/sgRNA依赖性和非依赖性脱靶效应。同时,该团队还表示,tBE系统在小鼠体内没有检测到全基因组和全转录组的脱靶效应。
2.1.2.6 降低序列背景依赖性 融合rAPOBEC1的BE3对TC序列编辑效率较高,但是对GC序列基本没有编辑效率[17];而融合了hAID、PmCDA1和hA3A系统则可对富含GC的序列进行有效编辑[33,64-65],且hA3A系统还能有效的应对高甲基化区域,这是其他编辑器不能做到的[66]。直到2018年,Li等[41]构建的dCpf1-BE(Cas12a)系统对其他编辑器做了有效的补充,该系统可以在AT富集(AT-rich)PAM序列区域实现有效的编辑。
2.2 腺嘌呤碱基编辑器ABE
2.2.1 ABE的建立 人类致病性的SNPs并非全部由T变异成C引起的,CBE碱基编辑器虽然可以实现C T(G A)碱基转换,但CBE系统仅能纠正部分变异,远远达不到纠正人类疾病的需求。因此科学家们迫切需要拓展出新的编辑器家族成员。基于与CBE系统同样的原理,David Liu实验室在2017年开发了可以实现A G(T C)转换的ABE碱基编辑器,ABE系统打破了编辑器只能编辑C或G的限制,从而扩大了碱基编辑的应用范围[18]。ABE系统的作用原理与CBE系统相似,在DNA复制过程中,腺嘌呤脱氨酶将靶位点处的腺嘌呤(A)经脱氨反应转变为肌苷(I),肌苷在DNA复制时会被错误当作鸟嘌呤(G)进行读码,因此,肌苷与鸟嘌呤互补的胞嘧啶(C)结合形成C-I碱基对,再经过一轮DNA复制,部分形成C-G碱基对,最终实现A-G的改变。ABE的开发并没有CBE系统那么一帆风顺,起初,David Liu实验室分别运用自然界中的大肠杆菌TadA、大鼠ADA、人类ADAR2和人类ADAT2四种腺嘌呤脱氨酶直接与nCas9融合,但几乎没有编辑活性[18]。这表明自然界中的腺嘌呤脱氨酶不能直接融合于碱基编辑器中。遂该团队决定采用将现有腺嘌呤脱氨酶改造的策略,以解除腺嘌呤脱氨酶在连接成单链或双链DNA时不能脱氨的限制。科学家们发现大肠杆菌埃希菌株的RNA腺苷脱氨酶TadA能够作用于多核苷酸且不需要额外的激活程序,遂以TadA作为改造对象对其进行人工随机改造。将随机突变的TadA序列与dCas9蛋白融合,构建成随机突变文库,再结合易错PCR、DNA重组等定向进化策略,再通过相应的抗生素筛选和测试,先后共经过7轮的改造和筛选,最终成功改造出了能直接作用于ssDNA的ABE7.10系统。在哺育动物细胞测试中,A G(T C)的平均编辑效率显著提高,由最初的3%提高到了58%,提高了约19倍,编辑活性窗口可覆盖sgRNA的第4-9位[18]。
2.2.2 ABE的优化 研究表明,自然界中缺乏可以直接起作用的腺嘌呤脱氨酶,因此,ABE系统的优化相对于CBE系统比较局限,其优化策略主要是建立在TadA7.10的基础上。又由于研究人员发现ABE系统无论是在编辑哺乳动物或者植物,相比CBE系统,ABE存在更高的准确性和更少的Indels,因此,ABE系统的优化主要表现在如何提高碱基的编辑效率、如何扩大编辑活性窗口和碱基编辑范围方面[11,67]。David Liu实验室将SV40 NLS换成bipartite NLS(bpNLS),并在TadA7.10的N端增加1个bp NLS,优化了其密码子,构建出了ABEmax,其编辑效率相较于ABE7.10进一步稳定提高,在HEK293T细胞中实验结果显示,ABEmax的编辑效率提高了1-6.1倍[19]。David Liu实验室开发的CP-ABEmax(CP1012-ABEmax、CP1028-ABEmax、CP1041-ABEmax和CP1249-ABEmax)系列碱基编辑器在一定程度上将碱基编辑窗口从4-8位拓展到4-12位[39]。与CBE系统相似,将识别不同PAM序列的SpCas9变体融入ABEmax系统,即可拓展ABEmax在基因组的编辑范围。目前,融合在ABEmax系统中的不同SpCas9变体或不同来源的Cas9蛋白有VQR-SpCas9(PAM:NGA)、VRERSpCas9(PAM:NGCG)、VRQR-SpCas9(PAM:NGA)、SaCas9(PAM:NNGRRT)、SaKKH(PAM:NNNRRT)、ScCas9(PAM:NNGN)、xCas9(PAM:NG)、SpCas9-NG(PAM:NG)、SpG(PAM:NGN)和SpRY(PAM:NRN),这些蛋白极大扩大了ABE在动植物基因组中的可编辑范围[39, 45, 48-49, 68]。
以上的改造比较局限,均是在ABE7.10或ABEmax的基础上,未涉及对TadA脱氨酶的升级替换。2020年,David Liu实验室通过噬菌体辅助的非连续和连续进化(PANCE和PACE)系统改变了ABE7.10的脱氨酶成分,产生了具有Cas结构域兼容性与活性均得到提升的腺嘌呤碱基编辑器ABE8e[50]。与ABE7.10相比,ABE8e包含8个额外的突变,活性提高590倍。该团队还证明,ABE8e系统还可以编辑ABE7.10编辑效果不佳的靶点。但不幸的是,ABE8e的脱靶率比ABE7.10高,为了解决ABE8e高脱靶率的问题,该团队将TadA-V106W突变引入了ABE8e系统,创建了ABE8eV106W,大幅度降低了ABE8e在DNA和RNA上的脱靶效应[50]。Lapinaite等[69]对ABE8e进行了超高分辨率冷冻电镜结构分析,结果表明,最新的ABE8e对DNA的脱氨基速率比ABE7.10和miniABEmax分别高出590倍和1 170倍。2022年,赖良学团队在ABE8eV106W的基础上优化出可以识别具有NTTN、TYCN和TRTN形式PAM的ABE8e编辑器(enCas12a-、RR-和RVR- ABE8eV106W)[42]。超高的编辑效率和编辑范围大大增强了ABE碱基编辑的应用范围。
2.3 糖基化酶碱基编辑器GBE
在CBE行使功能的过程中[53],C变为U后,会被细胞内源的尿嘧啶-DNA糖基化酶(uracil-DNA glycosylase, UDG)酶水解,成为无嘌呤无嘧啶AP状态,进而引发碱基切除修复(base-excisionrepair,BER)机制,恢复为原来的C碱基,或者被AP裂解酶(APlyase)识别,引发DNA双链断裂,造成随机突变。但是不管发生哪种情况,都是在降低CBE编辑器的编辑效率,甚至引入随机突变。因此,在CBE系统的优化过程中,引入了外源的UGI(uracilglycosylaseinhibitor)序列来抑制UDG的活性,从而保护U碱基,提高C到U再到T的转换效率。DNA序列在AP状态下经过AP裂解酶作用缺失的碱基有几率被修复为其他3种碱基,为了保持AP状态,寻找C碱基转化为其他3种碱基的可能,就需要在CBE的基础上除去UGI序列,增强UDG序列的活性。由于GBE更加依赖于机体自带的修复机制,因此,C转变成为G或A存在不确定性和不稳定性。
CBE和ABE碱基编辑器的功能是实现同类碱基间的转换(嘌呤至嘌呤、嘧啶至嘧啶),但不能实现碱基间的颠换[70]。一些科学家另辟蹊径,以CBE系统产生的副产物为突破口,寻找其他的可能。2021年7月,美国哈佛医学院[53]和中国科学院天津工业生物技术研究所的张学礼研究员[20]相继在Nature Biotechnology上发表了新构建的碱基编辑器GBE,GBE能有效的将DNA靶点处的C-G及C-A碱基进行颠换修饰。2项研究采取了类似的开发思路,即GBE编辑器在CBE编辑器的基础上去除UGI的同时又增强UDG的功能,以期获得更高的C G或者C A颠换频率。David Liu团队使用CRISPRi(CRISPR干扰)技术针对碱基颠换过程中的影响因素(转氨酶和Cas效应结构域以及多种DNA修复蛋白)从而确定了影响C G至G C编辑结果的十多种序列特征[70]。并开发了与机器学习模型配套的系列工程化CGBE和预测文库CGBE-Hive。Yuan等[54]通过筛选不同来源的尿嘧啶DNA N-糖基化酶(uracil-N-glycosylase,UNG)和改变其与脱氨酶的种类和位置,结合密码子优化等策略,开发了效率更高的OPTI-CGBE。该团队通过内源位点及文库水平的研究并进一步开发出TCN PAM偏好性的eA3AOPTI-CGBEs,偏好CCN PAM的hA3G-OPTI-CGBEs和hA3G-CTD-OPTI-CGBEs等编辑器。
2.4 腺嘌呤碱基颠换编辑器AYBE
与GBE类似,一些科学家也在寻找嘌呤颠换为嘧啶的可能性。但是与CBE系统相比,ABE系统的副产物极少,因此开发AYBE的难度可想而知。科学家们发现N-甲基嘌呤DNA糖基化酶(N-methylpurine DNA glycosylase, MPG)有一定的切除肌苷(I)中次黄嘌呤碱基(Hx)的活性,于是猜想可以通过工程化改造MPG,以达到开发出可以将嘌呤颠换成嘧啶碱基编辑器的目的。令人兴奋的是,2023年1月,杨辉课题组与胥春龙课题组合作,率先实现了这一目标,开发出了新型可以颠换A成Y的碱基编辑器——腺嘌呤碱基颠换编辑器(AYBE,Y= C or T),首次实现了高效的腺嘌呤碱基颠换[21]。该系统通过在ABE8e的C端与野生的次黄嘌呤切除蛋白MPG融合,得到了最初的AYBEv0.1系统。为了提升AYBE的颠换效率,该团队对MPG序列开展了一系列的优化,并构建了一系列突变体库,经过多轮的筛选优化和测试,最终成功获得了碱基颠换效率较高的突变体AYBEv3。该研究显示,AYBEv3的颠换编辑效率是AYBEv0.1的4.78倍。而且AYBEv3和AYBEv0.1均有着相当的低Indels频率。
2.5 双重碱基编辑器DBE
2020年,高彩霞课题组率先开发出了双重碱基编辑器——饱和靶向内源基因突变碱基编辑器STEME(saturated targeted endogenous mutagenesis editors),拉开了双重碱基编辑器DBE的研究序幕[22]。该编辑器结构中包括可以实现双碱基编辑功能的元件有胞嘧啶脱氨酶APOBEC3A、腺嘌呤脱氨酶ecTadA-ecTadA7.10以及起引导作用的蛋白nCas9(D10A),依次通过48或32个氨基酸连接成为一个大的融合蛋白,并在nCas9的C端融合2个NLS拷贝和1个UGI拷贝。从而构建了STEME-1和STEME-NG两种编辑器。该研究还表明,创建的STEME系统可以在单个sgRNA的引导下在靶点位置同时产生C T(G A)和A G(T C)的突变。紧接着,Nature Biotechnology刊登了3篇采用类似策略的双碱基编辑器。Zhang等[23]将hAID、TadA与nCas9依次连接,并辅以优化nCas9密码子、增加双分型核定位信号(bpNLS),以及融合2个拷贝的UGI和优化linker序列获得了双碱基编辑器A&C-BEmax;结果表明,与CBE和ABE相比,A&C-BEmax编辑效率、产物纯度、可编辑窗口显著提高,而且几乎不产生DNA水平脱靶且RNA水平的脱靶也大幅降低。Sakata等[25]将胞嘧啶脱氨酶PmCDA1和腺嘌呤脱氨酶TadA与nCas9融合同样构建了双碱基编辑器Target-ACEmax。Target- ACEmax在47个被测试的基因组靶点上同样显示出较高的编辑活性。Grunewald等[24]则是将miniABEmax-V82G中的腺嘌呤脱氨酶和七鳃鳗中的胞嘧啶脱氨酶分别融合到nCas9的N端和C端,并添加两个拷贝的UGI,最终得到了双碱基编辑器SPACE。随后,该团队在HEK293T细胞中进行测试,数据显示,与miniABEmax-V82G和Target-AID相比,SPACE在靶点处实现了更加高效的双重编辑,且Indels产生率和脱靶概率也得到了有效降低。
2.6 引导编辑器PE
2.6.1 PE编辑器的建立 单碱基编辑器始终存在一定的局限性,不能随意转换4种碱基序列,对基因的插入和敲除也存在巨大的应用限制。2019年,David Liu团队在Nature上发表了新的研究成果,一种新型基因编辑技术——“prime editing”[26]。该技术直接将基因精准编辑推到了新的高度。PE系统的特点是可以直接靶向靶位点,在靶位点处进行基因精准插入、精准删除以及碱基修饰。最重要的是,PE系统有效避免了DNA双链断裂。PE系统的成功构建,大幅增加了基因精准编辑的编辑类型,同时,又大幅降低了基因编辑中脱靶效应的概率,提高了基因编辑的安全性和准确性。该编辑技术的核心之一是由逆转录酶和Cas9蛋白融合而成的特殊蛋白;另一个核心是在sgRNA的C末端增加了一段RNA序列,组成的特殊gRNA称为pegRNA。pegRNA具有双重作用,一段序列与sgRNA作用类似,作为靶点结合位点,与断裂的靶点处DNA链C末端融合以作为逆转录的起始位点,另一端序列则连接需要的目标逆转录序列。当PE行使功能时,nCas9蛋白在sgRNA的招募下,使DNA双螺旋打开,并在目标靶基因距离PAM序列上游3 nt处切割DNA单链。在游离的DNA单链处,DNA原序列与引物结合位点(primer binding site, PBS)结合,在逆转录病毒的作用下,以逆转录模板(reverse transcriptase templates,RTT)为模板开始逆转录过程,形成新的融合单链。最后经过细胞的修复机制和复制机制,有概率以新的DNA单链为模板进行复制,最终形成新的DNA双链,将目的基因整合到自身的基因组中(图2),以达到靶点突变、插入和敲除的目的。
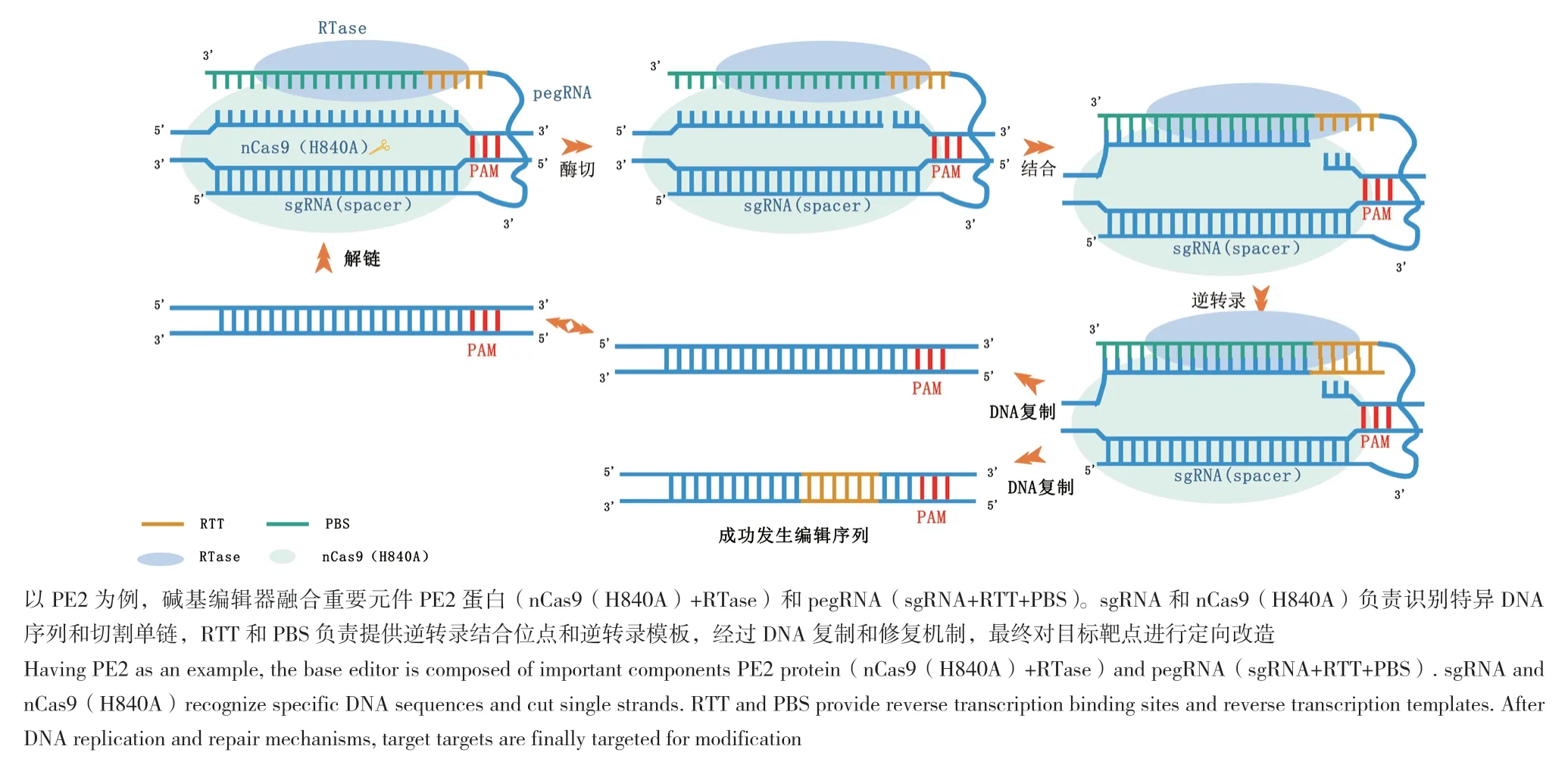
图2 引导编辑器工作原理示意图Fig. 2 Mechanisms of prime editors
相比于其他碱基编辑器,PE系统具有无可比拟的优势。(1)PE系统具有可编辑类型广的优势,包括实现所有的碱基突变和小片段的插入和删除以及他们的组合,这是其他碱基编辑器无法做到的;(2)较高的准确性,研究显示,虽然在PE3系统中检测出因为一条sgRNA的增加而增加了Indels产物,但Indels的水平大多在10%以下,仍然保持了较高的精准性;(3)高特异性,靶向区域DNA与pegRNA的spacer杂交、靶向区域DNA与pegRNA的PBS序列的杂交、靶向区域DNA与逆转录产物的杂交机制保证了PE系统的高特异性[26]。
2.6.2 PE编辑器的优化 PE编辑器虽然优势明显,但是,最初版本的PE编辑器编辑效率比较低,极大限制了PE的应用范围。得益于CBE(GBE)和ABE的优化改造经验,科学家们通过改造优化编辑器本体或融合新的辅助元件,对PE1编辑器进行了一系列的优化改造,将PE的应用范围大幅度提高。优化改造的方向包括提高PE的编辑效率、改变PE靶向PAM范围、提高目标产物纯度、降低副产物浓度等,且大部分改造策略已经被大量科学家证实可行并且产生了积极效果。
2.6.2.1 PE编辑器编辑效率的提升 PE1的编辑效率比较低[26],为了提高编辑效率,Anzalone等[26]对PE1的莫洛尼鼠白血病病毒逆转录酶(moloney murine leukemia virus reverse transcriptase,M-MLVRT)进行改造,在M-MLVRT酶中引入了5个突变位点构建了PE2,PE2逆转录酶的耐热性能、逆转录连续工作能力、催化DNA/RNA结合能力和对RNase的活性的抑制作用均得到有效提高,进而靶点处碱基转换、片段的插入或敲除效率得到提升(1.6-5.1倍)[26,71]。而随后Anzalone等[26]开发的PE3/PE3b进一步将编辑效率提高了1.5-4.2倍,而且,DSB和Indels也得到了有效的控制。Liu等[72]融合Csy4蛋白RNA加工系统而开发ePE系统平均提高4.9倍的编辑效率。Nelson等[73]的工程化pegRNA(epeg-RNA)则是在pegRNA的C端加入了防止C端降解的结构化RNA序列,测试发现,epegRNA的编辑效率提高了3-4倍。同样地,为了防止pegRNA的C端降解,Zhang等[74]和Li等[75]分别创制了xrPE和G-PE编辑器,同样将编辑效率提高了4倍。Li等[76]通过在RTT上引入同义突变和优化pegRNA二级结构的策略分别制备了spegRNA和apegRNA,二者大幅度提高了PE的编辑效率(353倍和2.77倍)。Lin等[77]采用双pegRNA的方式将编辑效率提高了17倍。Zhuang等[78]开发的HOPE系统显示出更高的编辑效率和产物纯度。融合了染色质调节肽(CMPs)、Rad51蛋白、c-Myc NLS、病毒核衣壳蛋白等功能性蛋白并结合优化密码子的方法已被证明是另一种有效的提高编辑效率的可行策略[73-77,79-86]。
2.6.2.2 PE编辑器可编辑范围的提升 与CBE(GBE)和ABE一样,PE编辑器的编辑范围严重依赖SpCas9蛋白。因此,扩大PE编辑器的应用范围就必须解除SpCas9蛋白对NGG形势PAM序列或编辑窗口的限制。目前,已经形成了可以识别多种PAM形式的PE编辑器变体,例如SpCas9-VQR(NGA)、SpCas9-VRQR(NGA)、SpCas9-NG(NG)、SpCas9-SpRY(NRN)、SaCas9(NNGRRT)[85,87]。此外,Oh等[88]构建的FnCas9(PAM:NGG)将SpCas9可编辑窗口扩大到PAM序列上游6-8 nt,大幅扩大了PE的应用范围。
2.6.2.3 PE编辑器敲除/敲入范围的提升 PE编辑器的功能异常强大,不仅可以定点修饰,还可以进行小片段的敲入和敲除,因此,PE受到广泛的关注。然而,在研究中,科学家们花费了大量的精力关注PE的突变功能,却少有人关注PE编辑器的敲除/敲入能力。值得兴奋的是,Anzalone等[89]通过采用构建两条相向的pegRNA替换PE编辑器内源性DNA序列的策略、成功制备了TwinPE系统;TwinPE系统可以实现108 bp片段的敲入和818 bp片段的敲除。基于同样的策略,Choi等[90]开发的PRIME-Del系统可以敲除长达10 kb的片段。Wang等[91]创制的GRAND则可以诱导150 bp序列敲入到目标基因组中,敲入效率甚至高达63.0%,且能够实现最长约1 kb序列的敲入。Tao等[92]的WT-PE系统采用靶向基因组上不同的位点,实现了长达16.8 Mb的大片段的敲除。这为敲除大片段DNA序列提供了新的思路。
3 新型碱基编辑器在猪中的应用
自2016年新型碱基编辑系统进入大众的视野,便迅速受到科学家们的青睐,并广泛应用于动植物及细菌的研究中。猪作为一种重要的农业经济动物,在农业和医学领域均具有重要应用价值,利用基因编辑技术能够快速提高猪的经济性状和模拟人类疾病。
3.1 新型碱基编辑器在农业中的应用
碱基编辑器可以通过提前引入终止密码子、调控内源mRNA的可变剪接和改变蛋白定向进化从而影响猪经济性状的表达。Wang等[93]利用碱基编辑技术(hA3A-BE3-NG)同时对猪的肌肉生长抑制素(myostatin, MSTN)、CD163受体、氨基肽酶-N(aminopeptidase, APN)以及MC4R四个基因进行编辑,产生了多个经济性状相关基因的无义突变和错义突变猪细胞。Pan等[94]则是通过rAPOBEC1-BE3编辑器在猪的MSTN基因中提前引入终止密码子,导致猪成纤维细胞中产生MSTN基因的无义突变。得到了MSTN基因缺失猪细胞。同样的策略,赵为民等[95]利用YE1-BE3-FNLS载体对猪的CD163基因第7外显子进行C到T的碱基突变,提前进入了终止密码子TAA,从而导致CD163基因不能完全翻译。王晶等[96]在宁乡花猪MSTN基因第2外显子处引入终止密码子,获得MSTN基因表达沉默的肾成纤维细胞系。王煜等[97]将rA1-BE3、hA3A-BE3、hA3A-BE3-Y130F和hA3A-eBE-Y130F4种胞嘧啶碱基编辑器分别作用于大白猪和巴马猪IGF2基因第3内含子第3 072 bp处的核苷酸,经细胞筛选后获得了C-T突变的猪细胞系。Song等[98]将hA3A中的第130位的酪氨酸突变到苯丙氨酸构建了hA3ABE3-Y130F体系,在猪胚胎成纤维细胞中的CD163、IGF2和MSTN基因的靶位点实现C-T精准修饰,导致无意或错义突变,生产出了即能抵抗猪蓝耳病病毒侵害,又显著提高了经济效益的仔猪。
3.2 新型碱基编辑器在疾病模型中的应用
在猪的疾病模型构建过程中,新型碱基编辑器的应用更加广泛。2018年,Li等[99]运用BE3系统对猪胚胎成纤维细胞的Twist2基因和Tyr基因做了定点修饰,并通过细胞核移植(somatic cell nuclear transfer, SCNT)方法建立了无脸巨口综合征和皮肤白化病的疾病模型。Xie等[100]运用BE3系统结合SCNT技术得到5头免疫缺陷症仔猪。该团队[100]运用同样的策略构建了400个克隆猪胚胎,并将克隆胚胎移植到代孕母猪,获得了1头健康的DMD单等位基因精准突变母猪。Zhu等[101]利用ABEmaxAW对猪胎儿成纤维细胞GHR基因可变剪接体做定向修饰,成功制备出4组纯合突变细胞。Yuan等[102]将BE4-Gam与GGTA1、B4galNT2和CMAH基因的3组sgRNA结合,结合后的3组sgRNA-BE4载体通过电穿孔法转染到巴马猪胎儿的成纤维细胞,结合SCNT技术,得到1只3基因精准修饰的可解决免疫排斥问题的仔猪。Yao等[103]以瓦登伯革氏综合征模型猪为基础,利用HA3A-eBE-Y130F对突变猪早期胚胎进行了干预。结果表明,通过基因编辑手段,可以将该病彻底纠正。但该编辑器产生了较为严重的脱靶效应,是HR途径的5倍,需要进一步的优化和降低脱靶效应。Jiang等[104]使用hA3ABE3-Y130F在大白猪胎儿成纤维细胞中过早引入终止密码子到3个肿瘤抑制基因TP53、PTEN和APC中。在290个分离的单克隆群落中得到232个三基因修饰群落。Zheng等[105]使用BE4max在猪睾丸干细胞中的猪内源性逆转录病毒(porcine endogenous retrovirus, PERVs)基因组上的gag(3个)和pol(4个)区域共设计7组sgRNA,在7个位点提前引入终止密码子以达到停止PERVs功能的目的,最终得到了少量的(10%)PERVs完全失活的单克隆。基因修饰后的干细胞与HEK293-GFP共培养的结果显示,完全失活的单克隆不能够整合到宿主细胞。
4 新型碱基编辑器存在的问题与展望
4.1 新型碱基编辑器的脱靶问题
单碱基编辑器作为重要的研究和改造单碱基变异的研究工具,其靶向特异性和脱靶效应受到了科研工作者们广泛而持续的关注。碱基编辑器导致的脱靶效应已经被大量科学家们相继报道。高彩霞组将碱基系统的脱靶分为传统的可预测的脱靶、全基因组范围内不可预测的脱靶和转录水平脱靶[106]。
基于传统的可预测的脱靶效应,一般可以通过软件进行脱靶位点的预测,如CRISPOR(http://crispor.tefor.net/),后续通过传统分子生物学的方法检测即可确定是否存在脱靶效应。David Liu实验室构建的高保真型BE3系统[43]、Lee等[63]筛选出的高特异性Sniper-Cas9蛋白、Wang等[44]开发的tBEs均可以有效的降低传统的脱靶效应。还可以通过改变sgRNA的长度、运用蛋白质工程的方式解决此类问题[43, 61-62]。
基于后两种脱靶问题,目前所开发的软件不能做出有效的预测,需要通过全基因组测序或者转录组测序获得[43,61]。2019年,Jin等[59]和Zuo等[60]分别在植物和动物中发现BE3存在全基因组的脱靶现象,而ABE在全基因组水平未检测出明显的脱靶现象,这表明ABE在全基因组水平上的安全性远超过CBE,这可能与人工定向进化的脱氨酶是否存在UGI等元件有关。在转录水平上,同年,Grünewald等[44]通过对人类细胞转录组测序发现,BE3和ABE系统在转录组水平存在严重的脱靶效应。随后,David Liu实验室也发现了ABEmax在编辑哺乳动物细胞系时发生了能够检测到的RNA脱靶效应[46]。针对这类问题,可以通过降低脱氨酶的活性,从而降低脱靶的概率;可以借鉴ABE的开发经验,通过蛋白质工程技术,如蛋白质的定向进化和从头设计,开发出新的脱氨酶,在不降低编辑器效率的前提下,提高特异性;还可以将新型编辑技术与传统基因编辑技术融合,开发新的编辑系统,以提高编辑的准确性[51-52]。2021年,Wang等[44]开发tBEs碱基编辑器可以对非靶点进行锁定,降低依赖性和非依赖性脱靶效应,同时,经在全基因组和全转录组水平的检测显示,未发现脱靶效应。2022年赖良学团队将TALE融入ABE/CBE系统而创建的新型碱基编辑器TaC9-ABE/CBE,在保留了碱基编辑器高效率编辑的同时又可以完全消除Cas9依赖性脱靶[51-52]。其他的脱氨酶突变体结合的碱基编辑器如BE3-R33A、BE3-R33A/K34A、ABEmaxAW、ABEmaxQW、ABE7.10D53E和ABE7.10F148A均可以同时兼具编辑活性和降低特异性[28,46-47]。
4.2 碱基编辑系统的未来
碱基编辑技术的创建为精准医疗和精准育种提供了重要的研究工具,碱基编辑技术以其低廉的价格和较低的门槛迅速受到科学家的青睐。经过多年的研发和优化,目前碱基编辑器家族成员也十分丰富,功能也越来越多样化。目前,碱基编辑系统已经能够实现嘌呤至嘌呤、嘌呤至嘧啶、嘧啶至嘧啶、嘧啶至嘌呤的转换,可以完成小片段的插入和敲除;碱基编辑系统的局限性也越来越小,现在科学家们已经开发出了近似无需PAM序列依赖的编辑器;编辑窗口也可以随着需要扩大或缩小。然而,碱基编辑器相比传统的HR途径,却存在着更高全基因组水平及RNA水平的脱靶效应,存在着潜在的危险性。因此,碱基编辑系统在技术及应用等层面仍需要潜心研发优化,以达到同时兼备高效性和安全性的目的。
尽管碱基编辑技术已经广泛应用于人类疾病研究、人类疾病模型的创建、动植物精准育种、动植物机制研究、微生物研究等领域,但碱基编辑技术存在的脱靶效应带来的潜在危害仍让人心存畏惧。目前,虽然科学家们结合人工智能,通过深度学习建立的物理预测模型可以有效的预测传统型脱靶效应,且已经展示出无可比拟的优势[70,107-108]。但是,全基因组水平的脱靶和转录组水平的脱靶却无法有效预测,通过后期的检测方法虽能检测出是否存在脱靶效应,但是存在一定的滞后性。因此,需要进一步将碱基编辑与机器学习结合,通过收集规模化数据,建立庞大的数据库,优化映射模型,结合高效的人工智能大数据信息处理系统,寻找出影响脱靶效应的影响因素,为以后基因编辑发展方向和人工定向蛋白质进化提供详实的数据支持。
5 总结
碱基编辑技术在基础研究和应用等方面充满生机,碱基编辑技术的问世会加速科学家探寻自然的奥秘,将进一步推动以农业、生物医药为代表的生命科学的发展。未来,随着基因组序列信息的不断挖掘和生物体机制不断地研究,隐藏在自然界中的奥秘会越来越多的展示在我们面前;另一方面,随着计算机技术的快速发展,人工设计精准医疗、精准育种也许会变成现实。
猜你喜欢
中国实用医药(2021年20期)2021-08-04
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
党的生活·党员电教与远程教育(2019年1期)2019-03-06
铁道通信信号(2018年7期)2018-08-29
中国医药指南(2018年3期)2018-03-23
电子设计工程(2015年4期)2015-02-27
电脑爱好者(2014年24期)2015-01-13
