
氮掺杂石墨烯负载单原子Zr 催化CO2 加氢的密度泛函理论研究
2024-01-04 11:57王炜泽胡延岗王广钊
分子催化 2023年5期
王炜泽 , 刘 璐 *, 胡延岗 , 王广钊
(1.江苏大学 能源与动力工程学院, 江苏 镇江 212013; 2.长江师范学院 电子信息工程学院 超常配位键工程与新材料技术重庆市重点实验室, 重庆 408100)
近年来, 随着工业及交通运输的迅速发展, 全球二氧化碳(CO2)排放量逐年增加, 并于2021 年再创排放量新高, 达到3.39×1010t, 若各国不进行有力的减排治理措施, 未来9 年内将耗尽剩余的“碳排放”预算, 这会对全球气候造成灾难性的影响[1].“碳达峰碳中和”已成全球学术界、工业界的热点问题, 研究机构和学者正在探索高效的CO2治理方式.在众多CO2的转化利用技术中, CO2催化加氢是理想的低碳技术之一, 以CO2为碳源, 将其转化为甲醇(CH3OH)、甲烷(CH4)、甲酸(HCOOH)等高附加值产品, 一方面可以治理二氧化碳, 减少温室气体的排放; 另一方面催化加氢后的产物具有高经济效益和环境效益.
负载金属催化剂是常见的加氢催化剂, 并已在工业上实现商业化应用.在众多催化剂载体材料中,石墨烯(Gr)因其优异的力学性能、高比表面积、高电导率及较低的成本而被认为是一种优秀的载体材料.但无缺陷的完美石墨烯是化学惰性的, 实验研究表明, 掺杂金属原子的方法可以有效调整石墨烯薄片的化学性质, 使其对CO2吸附增强, 并提高表面反应活性[2-3], 且石墨烯上的空位与金属原子结合足够稳定, 可用于化学反应[4].金属原子与周围配位原子的相互作用, 会在金属原子周围产生电荷积累, 这块区域表现出较高的催化活性[5].Esrafili 等[6]采用密度泛函方法(DFT)研究掺杂Ti 的石墨烯纳米片(Ti-Gr)氢化CO2的能力, 研究表明Ti 原子掺杂能够增加石墨烯的表面缺陷, 改善石墨烯表面反应活性, 同时加强催化剂稳定性.Sirijaraensre 等[7]采用密度泛函方法用石墨烯负载金属铜研究CO2氢化生成甲酸的催化反应路径, 研究表明石墨烯表面的Cu 对H2具有强吸附性, 为H2异裂提供反应位点,生成的Cu—H 易于和CO2完成后续的氢化.Esrafili 等[8]利用石墨烯为载体, 采用第一性原理研究金属Pt 及Ni 作为负载氢化CO2的催化活性, 由于Pt 原子中的正电荷较Ni 原子更多, 更显著地影响了表面的反应性能, 因此Pt 掺石墨烯具有更高的催化活性.Kumar 等[9]研究氮掺石墨烯负载铜原子催化剂将CO2氢化为甲醇的产量, 实验对比结果表明氮掺石墨烯负载铜原子催化剂的甲醇产量为吡啶N 掺杂石墨烯的2 倍, Cu 原子掺杂对于提升反应活性, 增加反应物产量具有正向作用.
此外, 调整载体的结构和组成对改良催化活性也是一种方法[10].由于N 原子比C 原子具有更高的电负性, 将N 引入配位环境中, 作为金属原子的配位原子[11], 可以改变电子态的空间和能量分布, 从而显著提高金属掺杂石墨烯薄片的表面活性[12].N 掺石墨烯主要以下3 种构型: 吡啶N, 石墨N, 吡咯N,研究表明, 吡啶N 构型最有利于金属原子的结合[13-14].Deerattrakul 等[14]采用水热还原的方法将N 掺杂在石墨烯中, 研究其对CO2加氢制甲醇产量的影响,研究表明在3 种N 掺杂石墨烯构型中, 吡啶N 掺杂石墨烯更有利于改善金属的分散, 能够促进H2的解离, 增强CO2的吸附, 从而促进甲醇的产生, 通过实验结果对比发现, N 掺杂石墨烯的甲醇产量是未掺杂石墨烯的4 倍.在N 掺杂石墨烯基础上, 进一步引入金属替代N 原子, 可以形成性能优良的单原子催化剂.Liu 等[15]采用第一性原理对PtN3-Gr 的研究表明, N 掺杂可以在石墨烯的费米能级附近引入局域缺陷态, 使锚定的Pt 原子的扩散与聚集更加困难, 从而有效提升掺杂Pt 原子的稳定性.Liu 等[16]发现FeN4-Gr 在CO2-CO 转化过程中表现出良好的性能, 且在吸附表面能促进CO2的活化和质子化.Cai 等[2]对多种过渡性金属(Sc、Ti、Cu、Zn 等)掺杂N4-Gr 进行了研究, 研究结果表明, N4-Gr 结构可以改善金属原子对CO2的吸附能力, 通过对比得出,Ti 原子掺杂N4-Gr 具有最强的吸附能力和最高的电荷转移能力.Wang 等[17]采用DFT 研究NiN3-Gr中Ni 结合位点不同对结构稳定性的影响, 研究表明Ni 被3 个吡啶N 原子包围的位置产生的吸附能最低, 吸附高度最小, 为最有利的吸附点.
研究发现, Zr 掺杂可显著提高石墨烯对气体分子的吸附, 学者在开发吸附材料、探测材料、储氢材料等方面进行了研究[18-19].然而, 目前对于Zr、N 共掺杂石墨烯的催化性能及其在CO2催化转化方面的研究尚为不足.我们利用密度泛函理论计算, 对比了CO2和H2在ZrNx掺杂石墨烯(ZrNx-Gr)上的吸附, 探讨ZrNx-Gr 催化CO2加氢制甲酸和CO 的反应机理.
1 计算方法及模型
采用Vienna Ab-initio Simulation Package (VASP)软件包[20-21], 基于广义梯度近似(GGA-PBE)[22]和投影增强波(PAW)[23]方法对ZrNx-Gr 体系进行计算.N3-Gr 和N4-Gr 是氮掺杂石墨烯的常见结构, 因此在石墨烯原胞基础上, 采用p(5×5)超胞建模, 构建了ZrN3-Gr 和ZrN4-Gr 体系.使用5×5×1 的Monkhorst-Pack 网格进行弛豫和态密度计算, 截断动能取520 eV, 表面结构弛豫收敛的判定标准为Hellmann-Feynman 力小于-0.2 eV/nm.利用baber 电荷来评价反应中的电子转移情况, 采用CI-NEB 方法搜索过渡态[24].
ZrN3-Gr 和ZrN4-Gr 的形成能计算公式如下:
其中Eform为形成能,EZrN3-Gr和EZrN4-Gr分别为ZrN3-Gr 和ZrN4-Gr 的总能量,μN为N2分子能量的一半,μC为石墨烯中单个碳原子的能量,EGr和EZr分别为石墨烯的能量和Zr 金属原胞中单个原子的能量.
将CO2等吸附物定义为X, X 在ZrNx-Gr 上的吸附能(Eads, eV)定义为
其中,EXC代表吸附物X 吸附在ZrNx-Gr 时复合界面的总能量,EC与EX分别代表掺杂ZrNx的石墨烯的能量和吸附物X 的能量.
吸附的焓变和吉布斯自由能的变化(25 ℃)采用如下公式计算:
其中,E0是静态计算得到的体系能量,H298和G298是25 ℃时体系的焓值和吉布斯自由能.
2 结果和讨论
2.1 H2 和CO2 在ZrN3-Gr 和ZrN4-Gr 表面的吸附
首先计算了ZrN3-Gr 和ZrN4-Gr 的形成能, 分别为4.19 和2.33 eV, 因此ZrN4-Gr 是更容易形成的结构.由于催化反应发生的第一步是反应物吸附在催化剂表面, 因此进一步比较了H2和CO2在ZrN3-Gr 和ZrN4-Gr 上的吸附情况.图1 和图2 分别为H2和CO2在ZrN3-Gr 和ZrN4-Gr 表面最稳定的吸附结构和差分电荷密度(EDD)图, 由H2和CO2吸附引起的吸附能、焓变、吉布斯自由能和电荷转移等数据如表1 所示.H2和CO2在ZrN3-Gr 和ZrN4-Gr 表面吸附的构型没有很大的差异.H2均平行于石墨烯表面在Zr 位点吸附, 吸附能分别为-0.49 eV (ZrN3-Gr)和-0.47 eV (ZrN4-Gr), 两个H 原子与Zr 形成几乎等距的化学键, H—H 键从自由态的0.074 分别增加到吸附态的0.088 nm (ZrN3-Gr)和0.082 nm (ZrN4-Gr).由EDD 图可见, H 原子之间的电荷密度略微减小, Zr 与两个H 原子之间的电荷密度明显增加, 说明H—H 之间相互作用减弱,Zr—H 之间相互作用增强.

表1 CO2 和H2 在ZrN3-Gr 和ZrN4-Gr 表面的吸附能、 焓变、 吉布斯自由能与电荷转移量Table 1 Adsorption energy, change in the enthalpy, Gibbs free energy and charge transfer of CO2 and H2 on ZrN3-Gr and ZrN4-Gr surface

图1 CO2 和H2 分子在ZrN3-Gr 上吸附的结构图、 EDD 图Fig.1 Optimized structure and EDD diagram of CO2 and H2 adsorption on ZrN3-Gr
CO2通过C 和O 原子形成Zr—C 键与Zr—O键吸附在Zr 上(图1(b)和图2(b)), 同时该O 原子与C 原子之间的C—O 键长增加, 由自由态的0.126 增加到吸附态的0.142 nm (ZrN3-Gr)和0.144 nm (ZrN4-Gr), 吸附能分别为-2.17 eV (ZrN3-Gr)和-1.66 eV (ZrN4-Gr).在EDD 图中, CO2中C 原子及靠近Zr 的O 原子与Zr 原子间电荷密度显著增加,说明CO2吸附时伴随有从表面发生的相对较大的净电荷转移, 并形成了Zr—C 与Zr—O 离子键, 同时, CO2中的C 与吸附在Zr 上的O 原子间的区域电荷密度减小, 说明C—O 键减弱, 与C—O 键长增加相符.
当H2和CO2共吸附时, CO2的吸附特性与其单独吸附时特性相同, H2相比于单独吸附远离Zr位点(图1(c)和图2(c)), 表明CO2在ZrN3-Gr 上发生氢化时, 优先吸附CO2.两种分子的共吸附能为-2.24 eV (ZrN3-Gr)和1.67 eV (ZrN4-Gr), 小于H2和CO2单独吸附的吸附能之和, 因此共吸附将削弱两者与表面之间的相互作用.表1 数据表明, 相较于单独吸附, 共吸附中H2的电荷转移量明显下降,ZrN3-Gr 由单独吸附的0.30 降低至共吸附的0.01 e,ZrN4-Gr 由单独吸附的0.17 降低至0.02 e.CO2的电荷转移量只有轻微的下降, ZrN3-Gr 中由1.10 下降至1.07 e, ZrN4-Gr 中由1.10 降低至0.99 e, 说明H2和CO2的相互作用对于H2影响更大.EDD 分析佐证了以上结果, ZrN3-Gr 上发生共吸附时, H2和CO2分子内部的电荷密度分布情况与其各自单独吸附时的电荷密度分布情况类似, 但两个H 原子与Zr 间的电荷密度增量相比于H2单独吸附时显著减小, 证明ZrN3-Gr 表面对H2的吸附被削弱; ZrN4-Gr 中H2单独吸附时的电荷密度增量相比于明显小于ZrN3-Gr, 共吸附时ZrN4-Gr 中CO2的电荷密度增量也有所减少, 说明H2和CO2与ZrN4-Gr 的结合弱于ZrN3-Gr.与文献中数据对比可知, 在ZrN3-Gr 表面, CO2单独吸附和共吸附吸附能显著高于CoN3-Gr、FeN3-Gr 和ZrN3-Gr 表面[12,20], 说明ZrN3-Gr 表面更利于CO2加氢反应的发生.
投影态密度(PDOS)分析如图3 所示, 其中(a)、(b)、(c)为ZrN3-Gr, (d)、(e)、(f)为ZrN4-Gr.对于H2的吸附(图3(a)和图3(d)), H2轨道在主要位于-8.32 eV (ZrN3-Gr)和-7.92 eV (ZrN4-Gr)处, 并在费米能级处存在态密度; 对于CO2的吸附(图3(b)和图3(e)), CO2轨道在主要位于-8.68、-5.97、-3.06、-0.57 eV (ZrN3-Gr)和-7.22、-4.72、-4.31、-1.60、1.31 eV (ZrN4-Gr)处.共吸附后(图3(c)和图3(f)),ZrN3-Gr 中, H2在-8.32 eV 处的能量峰值比其单独吸附时显著降低, 在-8.18、-8.79 和-5.94 eV 能量处产生了新峰; CO2在-8.32 eV 处产生新峰, 同时,在-5.97 eV 处的峰下降, 在-3.06 eV 处的峰上升.在ZrN4-Gr 中, H2单独吸附时在-7.92 eV 处的能量峰移至-8.59 eV 且峰值显著降低, 在-7.97 和-5.33 eV 处产生新峰; CO2的能量峰相比于单独吸附也整体左移, 在-7.92 eV 处产生新峰, 在-8.59 eV 处的峰值下降, 在-5.74 eV 处的峰值上升.在共吸附时, 两种结构的H2与CO2的轨道存在交叠, 说明共吸附后H2和CO2之间产生相互影响.ZrN3-Gr 上发生共吸附后, CO2轨道整体变化不大, 而H2轨道显著受到CO2的吸附影响, 说明CO2与Zr 的相互作用更强.H2吸附时Zr 的电荷转移量为1.86 e, CO2吸附和共吸附时的电荷转移量为2.21 e (表1), PDOS 图中H2吸附时Zr 的态密度明显高于CO2吸附和共吸附时的态密度, 两者保持一致.ZrN4-Gr 上发生共吸附后, H2和CO2的轨道均发生了偏移, 轨道出现明显的交叠, 说明共吸附时两者存在较强的相互作用.

图3 CO2 和H2 分子在ZrN3-Gr 和ZrN4-Gr 上吸附的PDOS 图Fig.3 PDOS diagram of CO2 and H2 adsorption on ZrN3-Gr and ZrN4-Gr
综上, 在ZrN4-Gr 和ZrN3-Gr 上吸附时的吸附结构相似, ZrN3-Gr 上各吸附质的吸附能均大于ZrN4-Gr, 25 ℃的焓变和吸附自由能也具有相同的趋势(表1), 说明CO2、H2与ZrN3-Gr 的相互作用更强, 更利于CO2加氢反应的发生.因此, 虽然ZrN4-Gr 的形成能更低, 但是CO2加氢反应在ZrN3-Gr 上发生的几率更高, 因此将ZrN3-Gr 作为进一步分析反应路径的表面.
2.2 CO2 加氢生成甲酸的反应路径
CO2加氢生成甲酸反应在MN3-Gr 表面通过HCOO*途径进行, 该途径又可分为顺式或反式HCOOH 两种路径, 经研究发现, 通过反式HCOOH生成甲酸的途径占优[25], 因此, 我们仅讨论CO2在ZrNx-Gr 表面通过HCOO*途径生成反式HCOOH的反应路径.
CO2在ZrN3-Gr 表面加氢生成甲酸的反应路径和能垒如图4(a)所示.对于CO2在ZrN3-Gr 上催化加氢制甲酸, 反应从初始态IS-1 开始, 此时CO2和H2共吸附在Zr 原子上.随着CO2中的C 原子与H2中的H1 原子的相互作用, H1 原子向C 原子靠近, 形成过渡态TS-1, 这一过程导致了Zr 原子和CO2中C 原子间Zr—C 键的断裂, 此时CO2与Zr原子间仅通过O2 原子连接, 同时, H2的氢原子间的H—H 键长从IS-1 的0.077 增加到TS-1 的0.100 nm.由IS-1 到TS-1 所需的能垒仅为0.19 eV.随后,TS-1 中的H 原子进一步靠近C 原子形成C—H 键,最终获得一个甲酸盐(HCOO*)中间体, 即中间态IM-1, HCOO 通过O2 原子吸附在Zr 原子上, 随后,甲基基团围绕Zr—O 键旋转并略微远离Zr 原子,使Zr—O 键键长略增, 与此同时, H2 原子开始脱附,键长由0.187 增加到0.221 nm, 形成TS-2, 该过程所需能垒为1.85 eV.之后TS-2 上的Zr—H 键断裂,H2 完成脱附, HCOO*基团绕Zr—O 键旋转的同时吸附H2 原子迁移靠近O1 原子, 最终形成O—H键, 形成终态FS-1, 最终甲酸从FS-1 上脱附得到最终产物, 总反应为吸热反应.
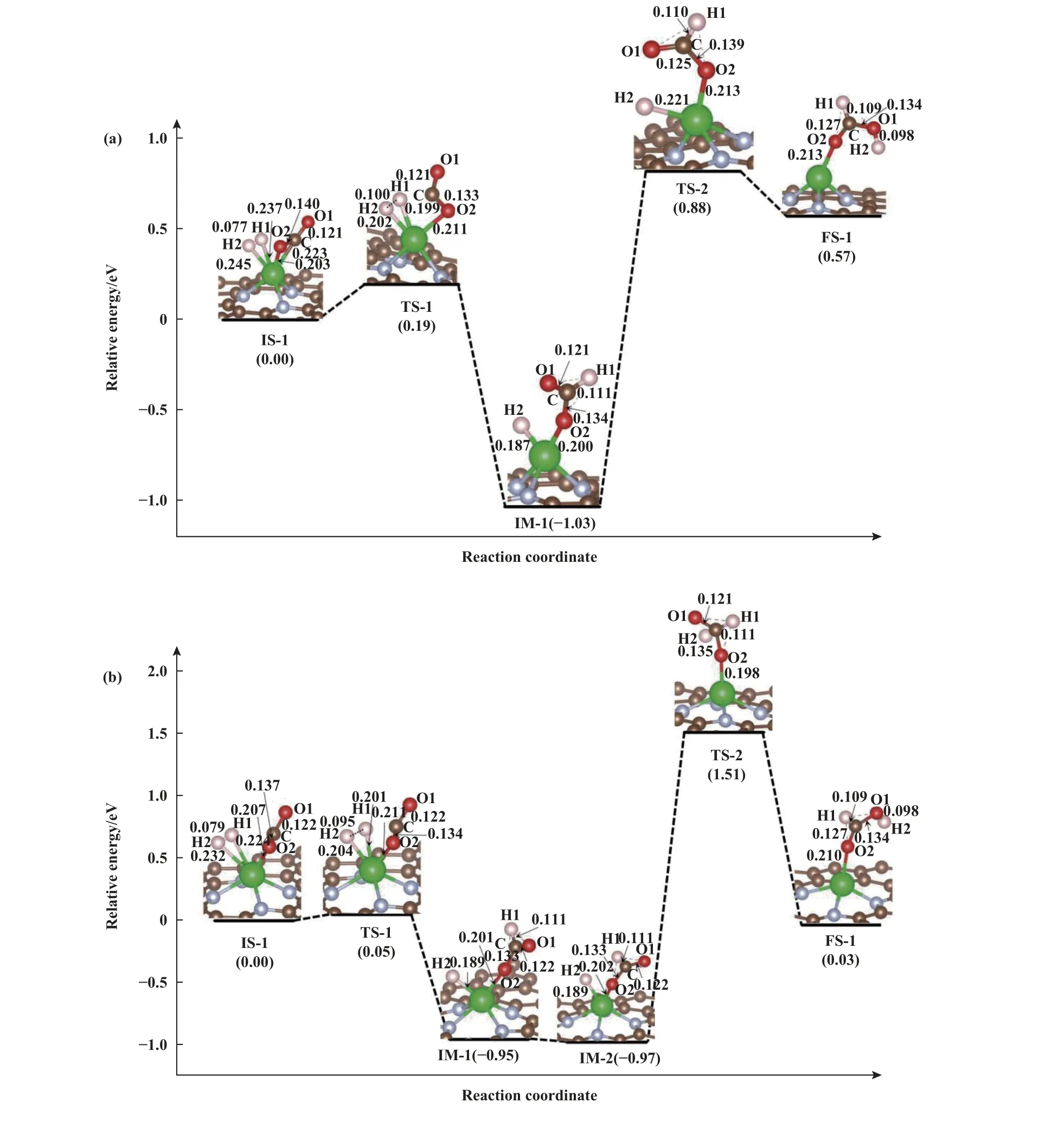
图4 CO2 在(a)ZrN3-Gr 和(b)ZrN4-Gr 上加氢制甲酸的路径Fig.4 The pathway of CO2 hydrogenation to formic acid on (a) ZrN3-Gr and (b) ZrN4-Gr
CO2在ZrN4-Gr 表面通过HCOO*途径生成反式HCOOH 的反应路径与在ZrN3-Gr 表面相似(图4(b)), 但在ZrN4-Gr 表面的反应路径中增加了IM-2 的中间态, 该构型由IM-1 上的HCOO*中间体经过一定的空间旋转所得.CO2在ZrN4-Gr 表面加氢生成HCOOH 决速步骤的能垒为2.48 eV, 高于在ZrN3-Gr 表面反应的能垒, 因此ZrN3-Gr 更利于催化CO2加氢生成甲酸.
2.3 生成CO 与H2O 的反应路径
CO2加氢生成CO 与H2O 的反应(CO2+H2→H2O+CO)是加氢制甲酸的一个重要的竞争反应[26], 该反应在ZrN3-Gr 表面的反应路径和能垒如图5(a)所示.反应从CO2和H2共吸附在Zr 原子上的初始态IS-1*开始, 经H—H 键, C—O 键的断裂与O—H 的生成形成过渡态TS-1*, 随即形成含OH*与CO*基团的中间态IM-1*, 其中TS-1*与IM-1*两者间键能等相差很小, 仅发生空间结构变化, 随后, H2 原子从Zr 原子脱离, 转移到O1 原子上形成过渡态TS-2*, 该过程所需能垒高达1.86 eV, 略大于CO2加氢制甲酸所需能垒.因此, 在ZrN3-Gr 表面进行催化加氢反应可分两条路径, 即HCOO*途径和CO 途径.

图5 CO2 在(a)ZrN3-Gr 和(b)ZrN4-Gr 上加氢产CO 与H2O 的路径Fig.5 The pathway of CO2 hydrogenation to produce CO and H2O on (a) ZrN3-Gr and (b) ZrN4-Gr
在ZrN4-Gr 表面的反应路径如图5(b)所示, 其反应决速步骤的能垒为1.73 eV, 小于在ZrN3-Gr 表面的反应决速步骤, 表明在ZrN4-Gr 表面更有利于该副反应的发生.
3 结论
通过密度泛函理论研究了氮掺杂石墨烯负载单原子Zr 催化CO2加氢的机理, 得到了H2和CO2在ZrN3-Gr 与ZrN4-Gr 上单独吸附及共吸附3 种状态下的吸附结构、 吸附能及电子转移情况.CO2在ZrNx-Gr 上加氢反应路径以H2和CO2共吸附的结构为起始结构, 在ZrN3-Gr 表面的吸附能显著高于ZrN4-Gr, 说明ZrN3-Gr 表面利于CO2加氢反应的发生.CO2在共吸附后保持了其单独吸附时的特性, 削弱了H2分子的吸附.ZrNx-Gr 上的CO2加氢反应分为两条路径, 一条路径生成HCOOH, 另一条生成CO 与H2O, 其中ZrN3-Gr 和ZrN4-Gr 表面反式HCOOH 形成的能垒分别为1.85 和2.48 eV, 产生H2O 与CO 的反应能垒分别为1.86 和1.73 eV.因此, 相比于ZrN4-Gr, ZrN3-Gr 更有利于CO2加氢生成甲酸反应的发生, 而ZrN4-Gr 表面更利于CO 的产生.
猜你喜欢
无机化学学报(2023年2期)2023-02-27
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中国蜂业(2018年4期)2018-05-09
黑龙江工程学院学报(2016年5期)2016-11-12
当代化工研究(2016年6期)2016-03-20
物理化学学报(2015年7期)2015-12-30
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
影像科学与光化学(2014年3期)2014-03-11
