
HPLC波长切换法同时测定淡竹叶中3种成分的含量
2010-02-07 03:48郭晏华
中成药 2010年9期
郭 妍, 郭晏华
(辽宁中医药大学药学院,辽宁大连116600)
淡竹叶为禾本科草本植物淡竹叶(Lophatherum gracile Brongn)的干燥茎叶[1]。主产于湖南,浙江,安徽,四川等省,是一味清心火,除烦热,利小便的良药,对治疗热病口渴,心烦不安,口糜舌疮,牙龈肿痛,小便赤涩淋痛,颇有疗效[2]。淡竹叶中含有大量的黄酮,内酯,氨基酸和糖类等成分,其中的黄酮类化合物具有降脂,抗血栓,抗氧化,降糖等多种生理活性,近年来受到国内外学者的关注。本实验测定了淡竹叶药材中3种指标性成分的含量[3],并比较了不同来源药材质量优劣,为临床评价淡竹叶质量提供参考。
1 仪器与试药
1.1 仪器 Agilent 1100型四元泵高效液相色谱仪、SPD-10A紫外检测器、SHB-3循环水多用真空泵、UV-4802型紫外-可见分光光度计、SHIMADZU AY220型电子分析天平、HH-4数显恒温水浴锅。
1.2 试药 反式对香豆酸对照品由天津一方科技公司提供,香草酸对照品(批号:110776-200402),牡荆素(苷)(批号:111687-200501)均购于中国药品生物制品检定所。本研究收集了15个不同来源的淡竹叶药材,经辽宁中医药大学李峰教授鉴定均为正品药材。甲醇为色谱纯,冰乙酸等均为分析纯,水为重蒸馏水。其它试剂均为分析纯。
2 方法与结果
2.1 色谱条件的选择
采用 AgiLent EcLipse XDB-C18(4.6 mm×150 mm,5 μm)色谱柱对流动相的组成、配比、体积流量等因素进行分析,对检测波长进行选择,确定最佳色谱条件。
2.1.1 流动相系统的选择 精密吸取供试品溶液10 μL进样,分别以甲醇-3%冰乙酸水、乙腈-3%冰乙酸水、甲醇-0.5%磷酸水溶液等不同浓度、不同比例的流动相系统进行等度和梯度洗脱,比较不同洗脱条件的色谱图,选择分离效果好,时间尽可能短的最佳色谱条件,最终确定以甲醇(A)-3%冰乙酸(B)为流动相进行梯度洗脱。流速:1.0 mL/min;柱温:30℃。见表1。

表1 流动相梯度洗脱程序
2.1.2 检测波长的选择 取一定浓度的香草酸,反式对香豆酸,牡荆素对照品置U-3010型紫外分光光度计下扫描,香草酸在260 nm有最大吸收,反式对香豆酸在308 nm有最大吸收,牡荆素在331 nm有最大吸收,为兼顾3种成分在同一色谱图上均有最大吸收,因此选用波长切换法。波长切换程序为:0 min→260 nm、30 min→308 nm、60 min→331 nm。
2.2 对照品溶液的制备
精密称取香草酸,反式对香豆酸,牡荆素对照品适量,分别置10 mL棕色量瓶中加甲醇溶解,制得浓度为0.276 mg/mL、0.223 mg/mL、0.231 mg/mL的对照品贮备液,备用。
2.3 供试品溶液的制备
精密称取淡竹叶药材(过40目筛)1.5 g,加入60%乙醇60 mL(pH=3),加热回流2 h,过滤。滤液蒸干,加热水15 mL,超声使溶解,离心10 min,转速为3 000 r/min。取处理好的大孔吸附树脂[4](AB-8)5 g装入柱中(1 cm×24 cm),离心液以2 BV/h流速上样(BV为树脂柱床体积),静吸附20 min。先用4 BV水冲至流出液无色,再用5 BV 20%乙醇冲洗至无色,然后用5 BV 40%乙醇冲洗至无色,最后用5 BV 60%乙醇冲洗至无色。最终收集40%和60%乙醇流出液,浓缩,用60%乙醇定容至10 mL量瓶中,经0.45 μm微孔滤膜过滤,滤液作为供试品溶液,备用。
2.4 系统适用性试验
精密吸取供试品溶液10 μL,注入高效液相色谱仪,记录色谱图,在选定的色谱条件下香草酸,反式对香豆酸和牡荆素的理论塔板数均不少于10 000,各成分与其他峰的分离度分别为 4.8,1.7,3.2。见图 1。
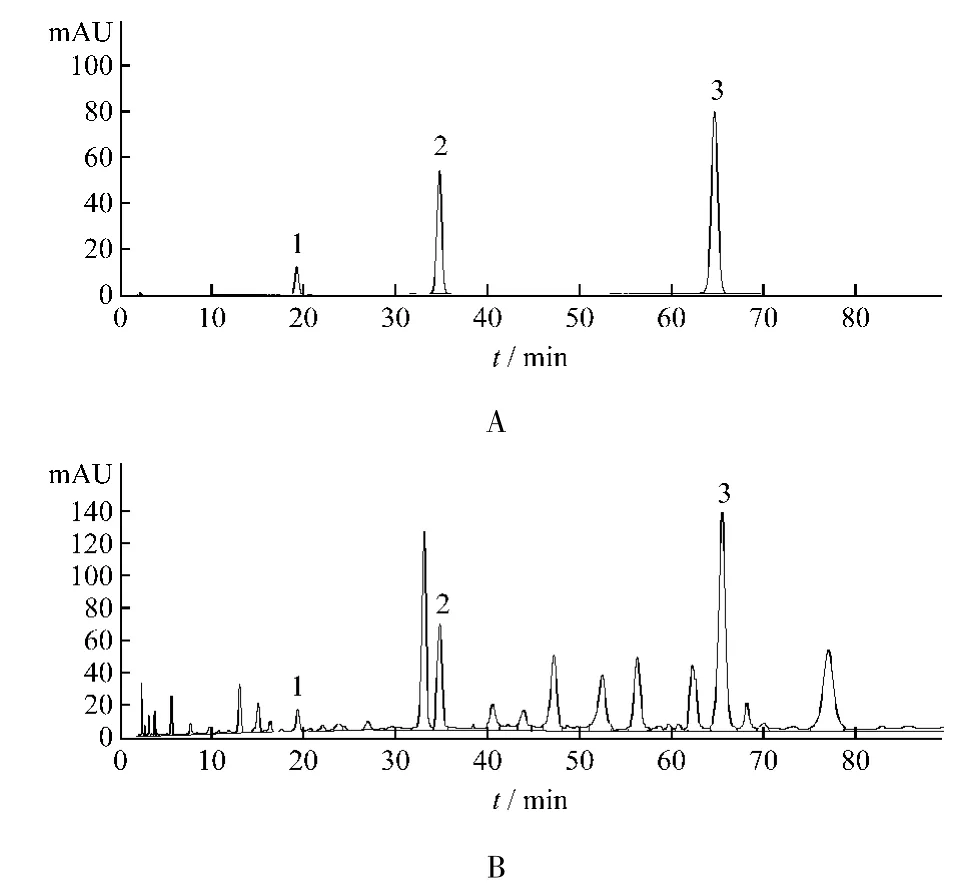
图1 混合对照品(A)和样品(B)HPLC谱图
2.5 方法学考察
2.5.1 标准曲线的制备 精密吸取香草酸对照品贮备液0.1 mL,反式对香豆酸对照品0.5 mL,牡荆素对照品3 mL置同一棕色5 mL量瓶中,加甲醇至刻度,摇匀,制得对照品混合液。精密吸取 4,8,12,16,20 μL 注入高效液相色谱仪,测定。以进样量(μg)为横坐标,峰面积为纵坐标进行线性回归,得香草酸回归方程为:Y=6 249.3X,r=0.999 3;反式对香豆酸回归方程为Y=9043.7X,r=0.999 8;牡荆素回归方程为Y=2 853.7X,r=0.999 9;结果表明:香草酸在0.022 08~0.110 4 μg范围内线性关系良好;反式对香豆酸在0.089 2~0.446 μg范围内线性关系良好;牡荆素在0.554 4~2.772 μg范围内线性关系良好。
2.5.2 精密度试验 取淡竹叶药材(1号)供试品溶液连续进样5次,记录色谱峰面积,结果香草酸、反式对香豆酸和牡荆素峰面积的RSD分别为2.2%、2.1%和2.8%。结论:结果表明,本方法精密度较好。
2.5.3 稳定性试验 取淡竹叶药材(1号)供试品溶液在0、2、4、8、12 h分别进样分析,记录色谱峰面积,结果香草酸,反式对香豆酸和牡荆素峰面积的RSD分别为2.9%、2.0%和2.5%。结论:结果表明,样品在本实验条件下稳定性良好。
2.5.4 重复性试验 取同一批淡竹叶药材(1号)样品5份,按供试品制备方法制备供试品溶液,并进样分析,结果RSD为香草酸2.9%,反式对香豆酸2.2%和牡荆素2.2%。结论:结果表明,本方法重复性较好。
2.5.5 回收率试验 取已知含量的药材(1号)5份,每份约0.75 g(香草酸含量0.040 17 mg/g,反式对香豆酸0.2084 mg/g,牡荆素1.532 mg/g)精密称定,置圆底烧瓶中,精密加入香草酸对照品1 mL(浓度为0.0276 mg/mL)、反式对香豆酸7 mL(浓度为0.0223 mg/mL)和牡荆素(浓度为0.231 mg/mL)5 mL,照供试品溶液制备方法制备。求得平均回收率分别为100.3%、100.2%和99.0%;RSD分别为2.5%、2.2%、1.4%。结论:结果表明,本方法回收率较高。
2.6 淡竹叶药材中香草酸,反式对香豆酸和牡荆素的含量测定
取不同来源的淡竹叶药材粉末(过40目筛)各约1.5 g,精密称定,制备供试品溶液,吸取各溶液10 μL,注入高效液相色谱仪。每个样品平行测定5次,计算各成分含量平均值及RSD。结果见表2。
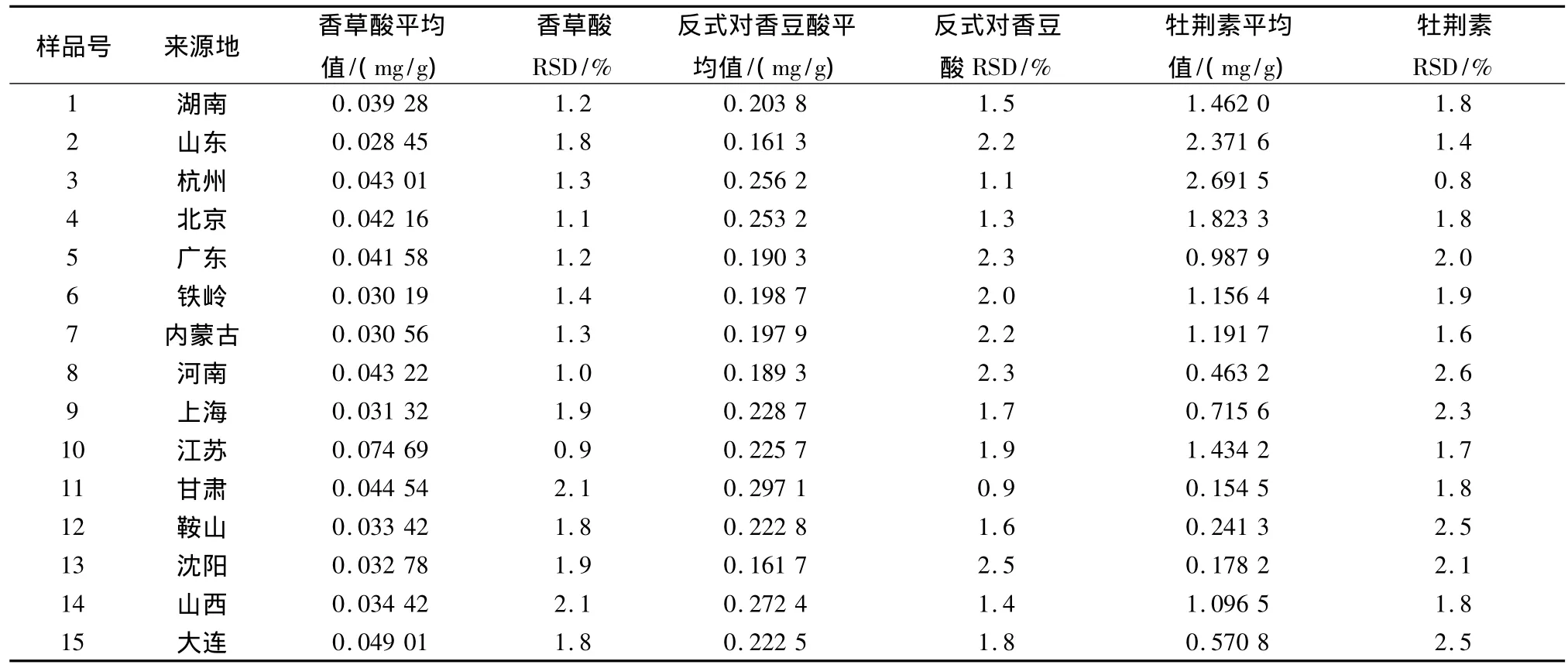
表2 不同来源淡竹叶中3种成分的含量
3 讨论
3.1 本实验首次采用高效液相色谱法同时测定了淡竹叶药材中香草酸、反式对香豆酸、牡荆素3种指标性成分,更好地控制了药材的质量,并且实验方法准确可靠,灵敏度高、重现性好,为淡竹叶药材的质量评价提供了有价值的参考。
3.2 通过对照品贮备液在200~400 nm进行紫外波长扫描,得出各自的紫外吸收曲线,根据相应的出峰时间确定波长切换梯度,使3种成分均在最大吸收波长测定,使测定结果更加灵敏、准确。
3.3 本实验比较了超声提取、水浴回流提取和索式提取的提取效果,最终确定水浴加热回流是最佳提取方法。同时通过正交设计实验考察了提取溶剂,提取时间,溶剂用量和提取次数对提取效果的影响。结果表明,以60%乙醇、60 mL、加热回流2 h提取效果最好。验证了提取时间过长使牡荆素(苷)降解,提取含量并不高。考虑到3种成分均偏酸性,因此加入盐酸调pH=3,使3种成分充分游离出来,便于提取完全。
3.4 淡竹叶药材中含较多如氨基酸,糖类等大极性成分,药材提取液杂质较多,直接测定会影响所测成分的分离度并且达不到基线,影响各成分的准确测定,因此采用大孔吸附树脂除杂。本实验考察了不同极性大孔吸附树脂处理效果,比较各经过处理的样品色谱图,最终选用AB-8大孔吸附树脂。该树脂能使淡竹叶药材提取液中的杂质除去,色谱图各峰之间分离度较好,达到基线分离,并且使所测3种成分损失较少。本实验考察了该树脂对淡竹叶药材提取液的吸附与解析能力,确定了最佳上样量、流速、所用洗脱溶剂乙醇的浓度梯度及洗脱溶剂体积。最终收集40%和60%乙醇流出液,经处理测定含量。
3.5 从表2可以看出不同来源的淡竹叶3种指标性成分含量差别很大,来自甘肃、上海、鞍山、沈阳的药材与其他商品地的相比含量较低。这可能由于是受生长环境不同所致。因此,淡竹叶应用于临床之前应对其质量进行评价。
[1]中国药典[S].一部.2005.
[2]宋秋烨,吴启南.中药淡竹叶的研究进展[J].中华中医药学刊,2007,25(3):526-527.
[3]陈 泉,吴立军,王 军,等.中药淡竹叶的化学成分研究[J].沈阳药科大学学报,2002,19(1):23-24.
[4]周 媛,程 凡.大孔树脂对葛根总黄酮的吸附及分离纯化研究[J].时珍国医国药,2007,18(12):3009.
猜你喜欢
南昌大学学报(理科版)(2022年6期)2022-02-04
开卷有益·求医问药(2021年7期)2021-07-22
中国生殖健康(2020年5期)2021-01-18
北方药学(2020年5期)2020-06-17
世界中医药(2019年2期)2019-09-10
中国生殖健康(2018年5期)2018-11-06
中国民族医药杂志(2016年5期)2016-05-09
中国现代中药(2015年6期)2015-09-25
中国现代中药(2014年7期)2014-11-02
中国药房(2013年7期)2013-12-03
