
β成核剂改性PPR/iPP共混物的力学性能与结晶结构的关系*
2011-03-15 08:43朱亚明刘述梅蒋文真周延辉赵建青
华南理工大学学报(自然科学版) 2011年6期
朱亚明 刘述梅† 蒋文真 周延辉 赵建青
(1.华南理工大学材料科学与工程学院,广东广州510640;2.广州市合诚化学有限公司,广东广州510620)
无规共聚聚丙烯(PPR)是丙烯和3.0%~7.0% (质量分数)的乙烯在一定温度、压力和催化剂作用下形成的共聚物,乙烯链节随机地分布到聚丙烯长链中,其主链结构规整性较等规聚丙烯(iPP)差,但它具有更优的抗冲击性能和耐压性能[1-2].与iPP一样,PPR仍为结晶型聚合物,晶型复杂,可为α、β、γ等晶型,且各晶型随加工条件的改变易相互转化[3].成核剂可较大程度地改变聚丙烯的结晶度和结晶形态,加快结晶速率.一般而言,α成核剂使聚丙烯结晶度增大,刚性增加,冲击强度则呈下降趋势;β成核剂使聚丙烯中β晶含量大幅提高,其独特的束状聚集结构在受力时产生裂纹带,可使韧性增加1倍以上,同时对其它性能没有明显的不利影响,因而β晶是一种最有应用价值的晶型[4-5].β晶在iPP结晶中的含量远低于α晶,熔点比α晶低20℃左右,有序程度和热力学稳定性方面低于α晶.γ晶则多由链缺陷或高压条件所致[6],Turner-Jones[7]早在1971年就发现iPP分子链上共聚乙烯会促进γ晶的生成,Mezghani、Laihonen等[8-9]的研究表明,乙烯含量的增加和较低的冷却速率有利于γ晶的形成.
各种成核剂对iPP的晶型改性一直是研究热点,冯嘉春等[10]研究发现,0.5%(质量分数)的镧配合物对iPP具有明显的α晶成核促进作用,并利用Avrami方程和Lauritzen-Hoffman对其结晶动力学进行研究发现,成核剂通过在更高的温度下增加晶核数目以及降低结晶生长时大分子在垂直于分子链方向折叠的界面自由能,使大分子链更易排入晶格;他们还发现在0.08%(质量分数,余同)的稀土配合物(WBG)或0.06%的N,N'-二环己基对苯二甲酰胺成核作用下,iPP体系β晶相对含量可达92%[11]. PPR工业化时间较短,有关其成核规律的研究相对较少,Xiao等[3]对WBG成核剂作用下PPR和iPP晶型的转变进行了比较研究.PPR与iPP分子链结构几乎相同,两者相容性好,有关各种成核剂对PPR,尤其是iPP的成核作用的研究较多,但对两者共混物的成核规律研究却未见报道.文中主要研究稀土配合物成核剂对PPR/iPP共混物的力学性能、结晶熔融行为的影响,探索其性能与晶型之间的相互影响规律.
1 实验
1.1 主要原料
PPR:牌号为R200P,熔体流动速率(MFR)为每10min 0.4g(190℃,5.0 kg),密度为0.91 g/cm3,韩国晓星厂家生产.iPP:牌号为basellHP550J,MFR为每10min 3.2g(190℃,5.0kg),密度为0.91g/cm3,韩国大林公司生产.β成核剂:稀土配合物,由中国科学院广州化学有限公司提供.
1.2 设备及仪器
SJSH-Z-30型双螺杆挤出机,南京橡塑机械厂; TTI-160F型塑料注射机,东华机械有限公司.
1.3 样品的制备
将PPR、iPP在70℃下干燥至恒重,然后与β成核剂等高速混合后进双螺杆挤出机熔融挤出造粒,挤出机各段温度设为175~195℃,主机转速为100r/min,喂料转速为40r/min.将挤出粒料充分干燥后,用注塑机制成标准试样,注塑压力为8 MPa,注射温度为180~200℃.
1.4 性能测试
拉伸性能按GB/T1040—1992进行测试,拉伸速率为50mm/min;冲击性能按GB/T1843—1996进行测试;维卡软化点温度按GB/T1633—2000进行测定.
采用NETZSCH DSC 200 PC型差示扫描量热仪(DSC),在N2氛围下从20℃以10℃/min的速率升温到220℃(第1次升温扫描),保温3min,以10℃/min的速率降温至20℃,然后再从20℃以10℃/min的速率升温到220℃(第2次升温扫描),再降温冷却,考察样品的熔融行为.采用D/max-IIIA型全自动X射线衍射仪(XRD)对注射成型的2 cm×2 cm试样在5°~40°范围内以2°/min的速度作步进扫描.采用日本JEOL公司JSM-6380LA型扫描电镜(SEM)对镀金后的冲击断面进行观察.
2 结果与讨论
2.1 成核剂对PPR/iPP共混物性能的影响
采用0.05%和0.20%(质量分数)的成核剂分别改性PPR、PPR/iPP共混物(质量比分别为70/ 30、50/50、30/70,分别记为70PPR、50PPR、30PPR)和iPP,对所得体系的力学性能和维卡软化点温度进行测定.成核剂用量为0.05%时,5个体系的Izod缺口冲击强度分别从42.7、33.7、14.5、8.3、5.1kJ/m2提高至87.8、63.4、29.3、13.9、8.2 kJ/m2,分别为添加前的2.1、1.9、2.0、1.7、1.6倍,维卡软化点温度分别从132.0、137.4、138.2、142.5、153.0℃下降至122.7、134.3、136.5、141.0、145.9℃,而拉伸强度下降幅度则均在10%以内;成核剂用量增至0.20%时,Izod缺口冲击强度反而比0.05%用量下低,分别为50.7、37.9、25.0、11.0、7.0 kJ/m2,略高于未加成核剂时,拉伸强度和维卡软化点温度则与未加成核剂时基本一致.以上结果说明:成核剂用量约为0.05%时,其对PPR、iPP及PPR/iPP共混物的增韧作用较大;成核剂用量增加到0.20%时,增韧作用反而降低.由于PPR的韧性远高于iPP,共混物的性能受PPR/iPP质量比的影响较大,两种成核剂用量下,仍是以PPR为基体的70PPR的韧性较好,以iPP为基体的30PPR的刚性较好,50PPR则介于两者之间.
2.2 DSC分析
iPP结构规整,结晶速率较慢,容易产生α晶,而PPR分子链中作为缺陷存在的乙烯链节使规整结构的聚丙烯链段运动变得相对容易,结晶性能发生改变,结晶速率较快,更容易产生β晶.PPR/iPP共混物的力学性能和热变形温度的变化与成核剂加入引起的结晶度和晶型的变化密切相关,文中采用DSC法对各体系的结晶与熔融过程进行研究.
PPR与iPP第1、第2次的DSC升温熔融曲线如图1所示.从PPR的谱图上可以看到:第1次升温扫描时,仅在167.1℃出现了一个典型的α晶(一般规整结构聚丙烯α晶的熔点为165~175℃[12],属于单斜晶系)熔融峰;而在第2次升温扫描图上,于151.7、165.7℃处出现熔融双峰,前者对应β晶(β晶熔点为143.0~157.0℃,此温度范围与γ晶和部分不完善的α晶熔点重叠,属于六方晶系)的熔点,相应的峰面积达到了66%,即重结晶后β晶含量大幅提高,后者归属于α晶,与第1次相比,其对应峰面积大幅度降低.两次升温扫描曲线所呈现的较大差别可能是由于:(1)结晶条件不同,第1次升温熔融反映注塑过程中的结晶状况,熔体经过复杂的热和流动环境,在模具中经循环水冷却20s后置于室温,过冷温度高,与生成β晶的理想温度110~130℃[6]相距甚远,因此试样中β晶含量偏低;而DSC测试中PPR以10℃/min加热,并在220℃保持3 min,得以充分熔融,然后以10℃/min降温,较慢的降温速率适宜β晶成长[13-14];(2)β晶在有序程度和热力学稳定性方面低于α晶,注塑制样过程中形成的较高含量的β晶在放置时转变成了α晶,或第1次升温熔融过程中发生了β晶向α晶的转变,致使第1次扫描只出现α晶的峰.鉴于测试样条良好的冲击韧性,第2个因素可能是主要原因.iPP第1、第2次升温扫描均表现为熔融单峰,分别在171.1、169.0℃处,峰温略有移动,均为典型的α晶熔融峰.PPR的α晶熔点比iPP低3.3℃,这是由于其分子链上存在丙烯与乙烯共聚单元,结构规整性降低的缘故,由此推断PPR的α晶熔点将降至162.0~172.0℃之间.

图1 PPR与iPP的DSC升温熔融曲线Fig.1 DSC melting curves of PPR and iPP
PPR和iPP中加入了0.05%的成核剂后(记为PPR+0.05%β和iPP+0.05%β)的第1、第2次DSC升温熔融曲线如图2所示.PPR+0.05%β在第1次升温扫描时于110.0℃附近出现较明显的肩峰,第2次升温扫描时该峰消失.这种现象可能与规整聚丙烯分子上作为缺陷存在的乙烯链段引起的γ晶有关[15],γ晶容易在高压条件下产生,本样条注射成型时较大的压力促使了γ晶的形成;第2次加热扫描时高压条件解除,γ晶难以生成,因此该肩峰不会出现.PPR+0.05%β的第1、第2次升温扫描熔融峰均出现在147.0℃附近,表现为典型的β晶熔融峰.iPP+0.05%β的第1、第2次升温扫描过程中均出现熔融双峰,但两峰强弱变化明显,峰位置有所变化.第1次扫描,双峰出现在153.2、173.2℃处,后者较强;第2次扫描时双峰出现在155.9、167.3℃附近,两峰靠近,且峰面积基本相当,表明0.05%的成核剂可促进iPP形成较高含量的β晶,但从第1、第2次升温扫描表现的差异推断,iPP体系所形成的β晶稳定性较PPR体系差.由此可以看到,0.05%的成核剂可促进PPR形成大量稳定的β晶,这是其冲击性能成倍提高的根本原因;0.05%的成核剂对iPP体系β晶的促进作用相对较弱,形成的β晶不稳定,且其β晶熔点较PPR的高8.9℃,所表现的冲击韧性远低于PPR.

图2 PPR+0.05%β和iPP+0.05%β的DSC升温熔融曲线Fig.2 DSC melting curves of PPR+0.05%β and iPP+ 0.05%β

图3 PPR+0.20%β的DSC升温熔融曲线Fig.3 DSC melting curves of PPR+0.20%β
PPR中加入0.20%的成核剂(记为 PPR+ 0.20%β)后的第1、第2次升温扫描曲线如图3所示.两曲线均表现为熔融单峰,第2次扫描时峰温为154.5℃,仍以β晶为主,较加入0.05%的成核剂时往高温方向移动了7.5℃,这一方面可能是由成核剂用量增加,结晶速率加快,结晶完善程度增加引起的;另一方面可能是由一部分α晶的形成引起的.从两种用量下体系冲击性能的变化(PPR+0.05%β体系高于PPR+0.20%β体系)以及α晶受力作用过程中变形吸收能量的能力较β晶差推断,较多α晶的形成是导致峰温向高温方向移动的主要原因. iPP中加入0.20%的成核剂后第1、第2次升温扫描结果与加入0.05%的成核剂较相似,峰温仅向高温处略作移动.
对于70PPR、50PPR、30PPR 3个共混物,其第1、第2次升温扫描曲线均表现为一个明显的熔融峰,说明PPR与iPP相容性良好.鉴于两次升温扫描峰温位移较小,仅将三者的第2次升温扫描曲线进行比较,如图4所示.由图4中可以看出,三者的熔融峰温分别为161.9、164.5、166.4℃,且随iPP组分比例的增加而升高;仅70PPR体系熔程较宽,在147.7℃处产生β晶肩峰,表明体系中α、熔点略低于iPPβ晶共存;30PPR体系则为典型的α晶.

图4 70PPR、50PPR、30PPR共混物的第2次DSC升温熔融曲线Fig.4 The second DSC melting curves of 70PPR,50PPR and 30PPR blends
加入了0.05%和0.20%的成核剂的70PPR体系(记为70PPR+0.05%β和70PPR+0.20%β)第1次扫描时在110.0℃附近均出现γ晶肩峰,第2次扫描时该峰消失,第1、第2次升温扫描的其它谱图部分基本重合,均表现为明显的熔融单峰.两者的第2次升温熔融曲线与70PPR的比较如图5所示,峰温分别为 149.8℃和 159.8℃.根据峰温判断,0.05%的成核剂对70PPR共混体系的β晶成核促进作用十分明显,体系以β晶为主,且形成的β晶有序性高,十分稳定;成核剂用量增至0.20%时,熔融主峰温度提高至159.8℃,向α晶的熔点靠近,比未加成核剂的峰温低2.1℃,仅在146.5℃处出现一弱的β肩峰,表明成核剂用量超过一定值后,对β晶的成核促进作用降低,对α晶的促进作用相应增强.按附生成核机理[16],只有在一定浓度范围内的β成核剂作用下聚丙烯才能形成完善且数目较多的β晶,成核剂用量超过一定值后,结晶速率加快,附生中心来不及完善(即聚丙烯分子的手性调整不能满足β晶型形成的手性要求)而产生缺陷,导致β晶相对含量降低,而α晶相对含量相应增加.成核剂用量对70PPR的成核作用与对PPR的相似,冲击韧性的变化也是由成核剂引起的晶型变化所致的.

图5 70PPR、70PPR+0.05%β和70PPR+0.20%β体系的第2次DSC升温熔融曲线Fig.5 The second DSC melting curves of 70PPR,70PPR+ 0.05%β and 70PPR+0.20%β
由上述分析可知,成核剂用量为0.05%时,其对PPR、70PPR共混物的β晶促进作用较强,而用量增至0.20%时其对β晶的促进作用反而减弱,采用DSC法进一步研究0.20%成核剂对50PPR、30PPR两体系(记为50PPR+0.20%β和30PPR+0.20% β)的作用,它们第1、第2次的升温熔融曲线如图6所示.从图6中可以看到,第1次升温扫描两者均表现为双峰,峰温在152.0、166.0℃附近,前者归属于β晶,后者归属于α晶,后者强度远高于前者,以α晶为主;第2次升温扫描50PPR的两峰温为151.5、164.8℃,30PPR的两峰温为152.4、167.0℃,随iPP用量的增加两峰温均向高温方向即纯iPP的α、β晶熔点移动,按峰面积判断两者的 β晶含量,均在50%以上,说明有较强的β晶成核促进作用,但从两次扫描曲线的差异可以判断所生成的β晶的稳定性较差.从缺口冲击强度数据可知0.20%的成核剂的加入使50PPR、30PPR的冲击韧性增加,但增加幅度较0.05%用量时小,这同样是因为0.20%的成核剂在促进β晶的同时对α晶的促进作用加强了,体系内α晶的含量较高且结晶较完善,因而冲击韧性较0.05%用量时降低.
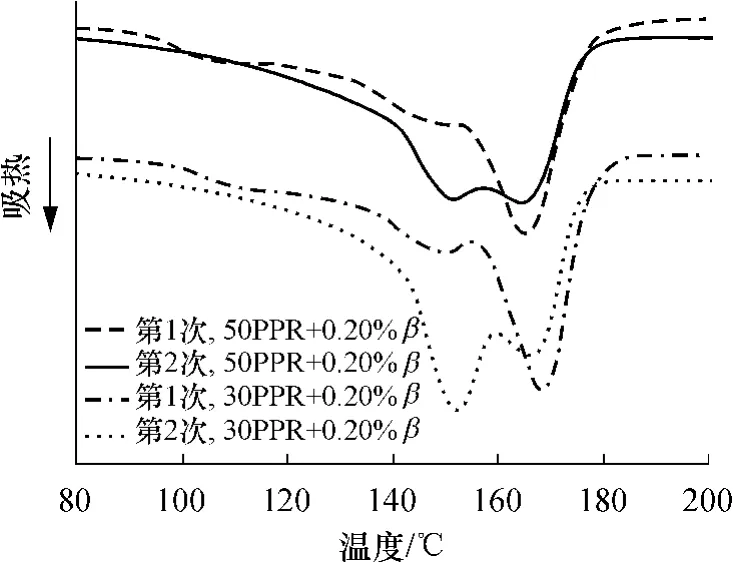
图6 50PPR+0.20%β和30PPR+0.20%β体系的DSC升温熔融曲线Fig.6 DSC melting curves of 50PPR+0.20%β and 30PPR+ 0.20%β
2.3 XRD分析
熔融过程中会发生β晶型向α晶型的转变,因此DSC升温曲线上各熔融吸热峰值并不能表征样品中各晶型的实际含量.XRD是在常温下对样品进行测定,测定过程中温度不发生变化,不存在各种晶型转变问题,因而可以较准确地得出各晶型的相对含量.
从DSC图的分析可知,70PPR体系中加入0.05%、0.20%的成核剂后,其第1、第2次扫描熔融曲线基本重合,晶型结构稳定,与PPR一同进行XRD分析的结果如图7所示.谱图上从左至右均出现2θ为16.3°、18.6°、19.5°、21.5°、24.4°和25.1°的峰,其中前三者分别为 β(300)、α(130)、γ(117)[8]晶的特征衍射峰,后三者分别归属于β(301)、β(130)和 α(060)[17].比较谱图可知16.3°、18.6°两峰的强弱随成核剂用量发生明显变化,19.5°峰的变化相对较小.以α、β、γ晶各峰面积占所有峰总面积的比值计算各晶的相对含量[18],结果如表1所示.

图7 PPR、70PPR、70PPR+0.05%β和70PPR+0.20%β的XRD谱图Fig.7 XRD spectra of PPR,70PPR,70PPR+0.05%β and 70PPR+0.20%β

表1 α、β、γ晶的相对含量Table 1 Relative contents of α-,β-and γ-form crystals
比较表中各数值可知,各体系均有较高含量的β晶、一定量的γ晶和较低含量的α晶,尤其是PPR本身结构中就含有较多的β晶.α晶的增强、β晶的增韧作用已得到广泛的认可,而γ晶对力学性能的影响还未见相关报道.PPR中β晶的相对含量最高,为56.1%,与30%iPP共混后,β晶相对含量降至45.3%,α晶的则增至12.4%.70PPR中加入0.05%的成核剂,β晶相对含量增至53.1%,与PPR相当,β晶成核作用明显,大幅提高的冲击韧性也表明了较多β晶的存在;当成核剂用量增加到0.20%时,α晶相对含量从0.05%用量下的11.3%增至15.1%,β晶相对含量则降至45.1%,α晶成核作用增强,β晶促进作用则减弱,这一变化说明了成核剂用量从0.05%增加到0.20%时,共混物冲击韧性的降低是由α晶相对含量的增加引起的.据此还可以推断,DSC谱图上两成核剂用量下熔融峰温从149.8℃移至159.8℃是由α晶的相对含量增加引起的.这里需要特别指出的是,谱图上19.5°所出现的强γ晶特征衍射峰在其它文献[8-9]PPR谱图上没有观察到,其对力学性能的影响还有待更深入的研究.
2.4 SEM分析
PPR、PPR+0.05%β、70PPR+0.05%β、70PPR+ 0.20%β体系的缺口冲击强度分别为42.7、87.8、63.4、37.9kJ/m2,韧性较高,对它们的缺口冲击断面进行SEM观察,结果如图8所示.从图8中可以看到:PPR冲击断面呈现一定的起伏,局部有少量塑性形变;PPR+0.05%β的断面可以观察到很多清晰排直的条纹状冲击剪切带,它们大致沿一个方向排布,高低不平且富有层次感,这是由β晶型独特的束状聚集体在材料受到外力时引发的,表现出了明显的韧性断裂特征;70PPR+0.05%β受到冲击时基体出现裂纹引发区和根部扩展区,裂纹扩展过程中呈现明显分支,这种裂纹耗散也能吸收较多的冲击能,表现为韧性断裂;70PPR+0.20%β同PPR冲击断面相似,呈现一定的脆性断裂特征.0.05%成核剂明显的增韧作用被很好地被反映在冲击断面上.

图8 样品缺口冲击断面的SEM照片Fig.8 SEM morphologies of Izod impact-fractured sections
3 结论
稀土配合物成核剂对不同质量比的PPR/iPP共混物均有一定的成核促进作用,其不同用量所引起的晶型和结晶度的变化也不同,导致所形成的PPR/iPP共混物的力学性能和热变形温度的变化规律有所差异.0.05%的稀土配合物将PPR、70PPR、50PPR、30PPR和iPP的Izod缺口冲击强度分别从42.7、33.7、14.5、8.3、5.1kJ/m2提高到87.8、63.4、29.3、13.9、8.2 kJ/m2;成核剂用量增加到0.20%时,分别为50.7、37.9、25.0、11.0、7.0 kJ/m2,冲击韧性反而降低.DSC、XRD研究发现,0.05%的成核剂对70PPR体系的β晶促进作用十分明显,成核剂用量增至0.20%时,对该共混物的α晶成核作用增强.缺口冲击断面的SEM观察表明,加入了0.05%成核剂的70PPR表现出明显的韧性断裂特征,加入了0.20%成核剂的70PPR的冲击断面则呈现一定的脆性断裂特征.稀土配合物成核剂用于50PPR、30PPR共混物所生成的β晶的稳定性较差,其作用规律还有待进一步研究.
[1] Hosier I L,Alamo R G,Esteso P,et al.Formation of the alpha and gamma polymorphs in random metallocene-propylene copolymers.Effect of concentration and type of comonomer[J].Macromolecules,2003,36(15):5623-5636.
[2] Stagnaro P,Costa G,Trefiletti V,et al.Thermal behavior,structure and morphology of propene/higher 1-olefin copolymers[J].Macromolecular Chemistry and Physics,2006,207(22):2128-2141.
[3] Xiao W C,Feng J C.Comparative investigation on crystallization conditions dependence of polymorphs composition for beta-nucleated propylene/ethylene copolymer and propylene homopolymer[J].Journal of Applied Polymer Science,2010,117(6):3247-3254.
[4] 付一政,安峰,曲静波,等.β晶型成核剂增韧改性聚丙烯及其共混物的力学性能与结晶行为[J].高分子材料科学与工程,2006,22(2):185-188. Fu Yi-zheng,An Feng,Qu Jing-bo,et al.Study on mechanical properties and crystallization behavior of polypropylene and their blending toughened by β crystalline from nucleation agent[J].Polymer Materials Science and Engineering,2006,22(2):185-188.
[5] Grein C.Toughness of neat,rubber modified and filled betanucleated polypropylene:from fundamentals to applications[J].Advances in Polymer Science,2005,188:43-104.
[6] Shi Q,Cai C L,Ke Z,et al.Effect of the nucleating agent 1,3:2,4-di(3,4-dimethylbenzylidene)sorbitol on the γ phase content of propylene/ethylene copolymer[J].European Polymer Journal,2008,44(7):2385-2391.
[7] Turner-Jones A.Development of the γ-crystal form in random copolymers of propylene and their analysis by DSC and X-ray methods[J].Polymer,1971,12(8):487-508.
[8] Mezghani K,Phillips P J.γ-phase in propylene copoly-mers at atmospheric pressure[J].Polymer,1995,36 (12):2407-2411.
[9] Laihonen S,Gedde U W,Werner P E,et al.Crystallization kinetics and morphology of poly(propylene-stat-ethylene) fractions[J].Polymer,1997,38(2):361-369.
[10] 冯嘉春,段瑜,焦瑛,等.镧配合物对等规聚丙烯等温结晶性能的影响[J].物理化学学报,2005,21(12): 1431-1435. Feng Jia-chun,Duan Yu,Jiao Ying,et al.Effect of lanthanum complex on isothermal crystallization kinetics of isotatic polypropylene[J].Acta Physico-Chimica Sinica,2005,21(12):1431-1435.
[11] Xiao W C,Wu P Y,Feng J C.Effect of β-nucleating agents on crystallization and melting behavior of isotactic polypropylene[J].Journal of Applied Polymer Science,2008,108(5):3370-3379.
[12] Lotz B.α and β phases of isotactic polypropyle-ne:a case of growth kinetics“phase reentrency”in polymer crystallization[J].Polymer,1998,39(19):4561-4567.
[13] Cermak R,Obadal M,Ponizil P,et al.Injection-moulded α-and β-polypropylenes:I.structure vs.processing parameters[J].European Polymer Journal,2005,41(8): 1838-1845.
[14] Cermak R,Obadal M,Ponizil P,et al.Injection-moulded α-and β-polypropylenes:II.tensile properties vs.processing parameters[J].European Polymer Journal,2006,42 (8):2185-2191.
[15] Na B,Lü R H,Xu W F,et al.Effect of nucleating duality on the formation of γ-phase in a β-nucleated isotactic polypropylene copolymer[J].Polymer International,2008,57(10):1128-1133.
[16] 张晓东,史观一.成核剂含量对β晶相聚丙烯结晶与熔融行为的影响[J].高分子学报,1992(3):294-298. Zhang Xiao-dong,Shi Guan-yi.Effect of β-nucleator content on the crystallization and melting behavior of βphase polypropylene[J].Acta Polymerica Sinica,1992 (3):294-298.
[17] Foresta T,Piccarolo S,Goldbeck-Wood G.Competition between α and γ phases in isotactic polypropylene:effects of ethylene content and nucleating agents at different cooling rates[J].Polymer,2001,42(3):1167-1176.
[18] Krache R,Benavente R,López-Majada J M,et al.Competition between α,β,and γ polymorphs in β-nucleated metallocenic isotactic polypropylene[J].Macromolecules,2007,40(19):6871-6878.
猜你喜欢
弹性体(2021年6期)2021-02-12
陶瓷学报(2020年2期)2020-10-27
含能材料(2020年2期)2020-02-19
中国特种设备安全(2019年2期)2019-04-22
中国塑料(2015年6期)2015-11-13
中国塑料(2015年8期)2015-10-14
中国塑料(2015年7期)2015-10-14
新疆钢铁(2015年3期)2015-02-20
固体火箭技术(2014年5期)2014-01-16
火炸药学报(2013年4期)2013-01-29
