
PuO3分子激发态的外场效应*
2011-07-24 09:49谢安东伍冬兰罗文浪周玲玲
中山大学学报(自然科学版)(中英文) 2011年1期
谢安东,伍冬兰,罗文浪,阮 文,周玲玲
(井冈山大学数理学院, 江西 吉安 343009)
钚作为武器和能源材料已经获得了极为重要的应用,但由于其特殊的放射性衰变和活泼的化学性质,一直难于发现高效长久的存储方式,因此其表面物理和化学长期以来都是研究的热点之一[1-7]。核武器库存期间核材料的腐蚀和老化的原因之一是钚的衰变问题,它涉及两个方面:钚及其化合物,如氢化物和氧化物的激发态的性质与变化;氦气泡的生成累集与影响。这些问题与外辐射场、自辐射场(内辐射场)相关。因此,本工作在文献[8-13]基础上,进一步研究外场对分子激发态的影响。
锕系元素的全电子计算需要大得几乎不可能的计算量,鉴于原子性质主要决定于价层电子,可应用有效原子实势(ECP)或相对论有效原子实势(RECP,Relativistic Effective Core Potential)理论[14]。Pu原子的基电子组态为(Rn)5f67s2,采用相对论有效原子实势(RECP)取代内层78个电子(1s22s22p63s23p63d104s24p64d104f145s25p65d10),价电子层含16个电子(6s26p65f67s2),应用文献[14]给出的RECP和收缩价电子基函数(7s6p2d4f)/[3s3p2d2f],即将(7s6p2d4f)收缩为(3s3p2d2f)基函数,对于O原子选用6-311+G*基组函数,首先,采用密度泛函(DFT)方法B3LYP在上述基组水平上对PuO3在分子轴方向外电场(-0.005~0.005 a.u.)作用下的基态几何结构进行了优化,然后,在同样的基组水平采用含时密度泛函(TDDFT)方法TD-B3LYP研究同样外电场作用下对其分子激发态的影响。
1 理论和计算方法
外电场作用下分子体系哈密顿量H为
H=H0+Hint
(1)
其中,H0为无外电场时的哈密顿;Hint为场与分子体系的相互作用哈密顿量。在偶极近似下,分子体系与外电场F的相互作用能为
Hint=-μ·F
(2)
μ为分子偶极矩。根据Grozema等提出的模型[15-16],在电场作用下的激发能Eexc与电场强度F、偶极矩和极化率的变化量Δμ和Δα满足关系式
(3)
其中,Eexc(0)为无外场下的激发能。本工作对Grimme的半经验方法加以推广[17-18],即将含时(TD)和密度泛函(DFT)进行结合以精确地计算激发能,并且在哈密顿量中加入了与电场有关的项[10,19]。
PuO3分子为C2V,按其标准坐标计算,Pu和3个O原子位于yz平面,沿z轴方向加上一系列有限的外场(-0.005~0.005 a.u.,约(-2.5~2.5)×108V/M),采用B3LYP/Gen方法,对全构型能量梯度优化PuO3分子结构(图1)。 在GAUSSIAN03程序中分子的哈密顿量中加入了Hint=-μ·F,μ为分子偶极矩矢量,F为外场矢量[20]。采用TD-B3LYP/Gen方法,计算有外场作用下PuO3的前5个激发态,全部计算在GAUSSIAN03软件包进行[21]。
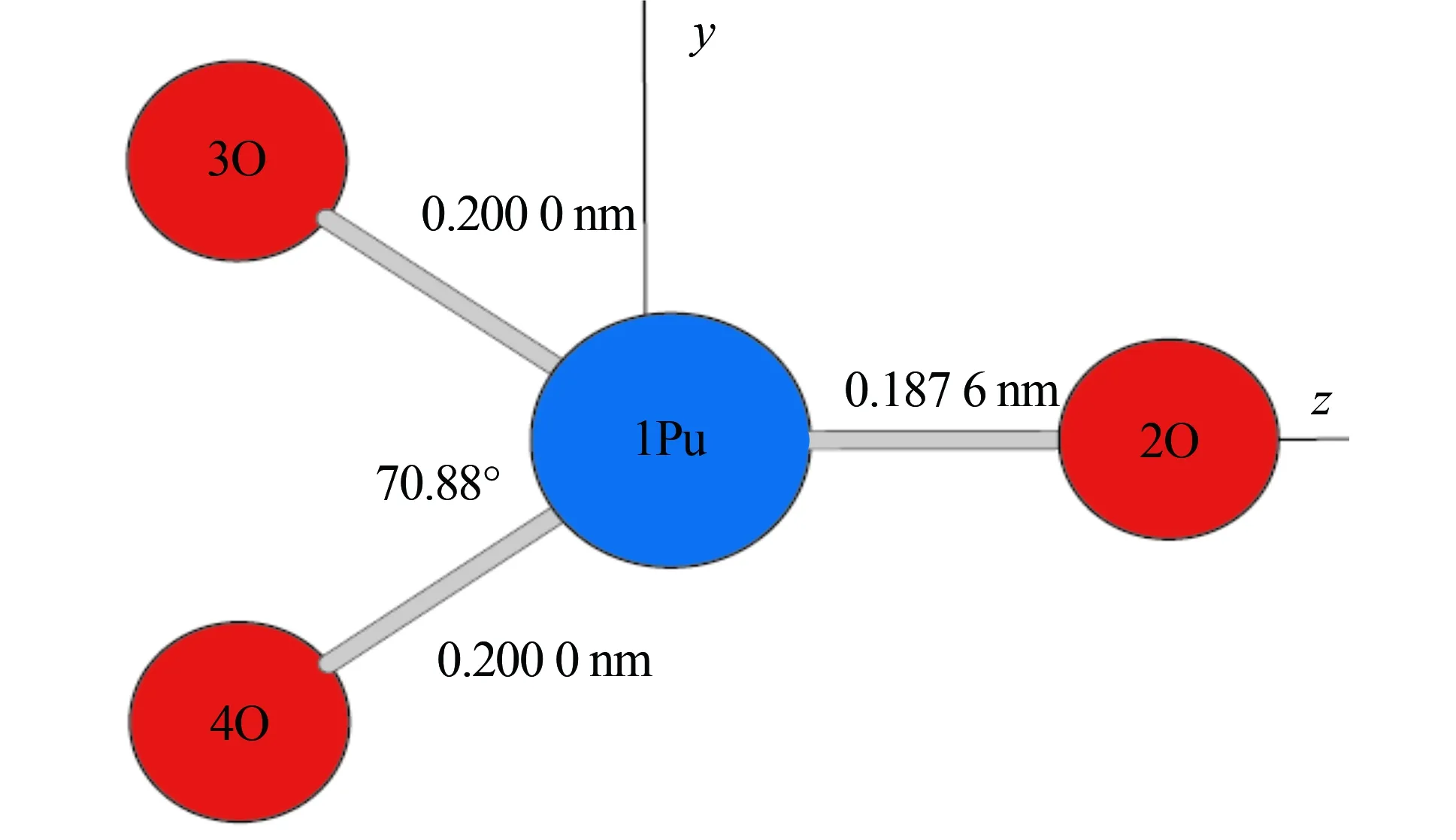
图1 无外场下优化的PuO3基态的分子结构
2 结果与讨论

由表1可以得出如下结论:
1)电场的引入改变了PuO3分子激发态出现的顺序。例如,F=0.000时,激发态顺序为7A1、7A1、7A2、7B2、7B1,而F=0.001 a.u.时,第2、3激发态发生交换7A1↔7A2,第5激发态由7B1变为7A1,F=0.003 a.u.时,7B2、7A1、7A2、7B2、7B1发生了更大的变化,其原因在于电场的引入改变了电子状态。
2)激发能随正向电场增大而减小,当反向电场增加时,激发能也是减小,如图2所示。激发能随电场变化的原因可根据前线轨道理论解释[22],例如,对第一激发态,当电场由-0.005变化到0.005a.u.时,分子最低空轨道LUMO与最高占据轨道HOMO能之差分别是3.553 9、3.560 9、3.572 4、3.579 5、3.581 03、3.585 31、3.565 3、3.550 6、3.484 2、3.476 6和3.469 5 eV,其定性规律与图2一致。

4)由激发能的计算可知,最高的激发能为1.377 3 eV,相应的波长为900.2 nm,最低的激发能为0.600 9 eV,相应的波长为2 063.3 nm。 PuO3的前5个激发态电子跃迁光谱波长为900.2~2 063.3 nm,属于红外-远红外光谱。
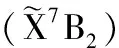
表1 PuO3从基态激发的前5个激发能E和振子强度f与外场F的关系1)
1)1.377 3 eV→900.2 nm,0.600 9 eV→2 063.3 nm,900.2~2 063.3 nm。

图2 激发能与电场的关系(1表示第1激发态;2表示第2激发态;3表示第3激发态;4表示第4激发态;5表示第5激发态)
3 结 论
1)由表1可知,在外场作用下对PuO3的前5个激发态电子跃迁光谱属于红外-远红外光谱,波长为900.2~2 063.3 nm;然而,对轻分子,如OH,NH,ZnF和H2O,电子跃迁光谱都在可见与紫外区,波长在63.98~594.97 nm.因为钚原子的基电子组态为KLMN5s25p65d106s26p65f67s2,有6个5f电子,处于离域和定域的转变间,所以5f电子有较大弥漫性,结合能比5d电子的弱,因而出现在红外-远红外区.这是钚原子的奇异特征。外场下PuO3的离解可能是首先由于Compton散射产生低能次级电子,然后再由次级电子碰撞使分子激发离解,分离出O,进而生成O2,O原子和O2分子又与Pu原子反应生成PuO、PuO2、PuO3,这样加速了Pu的腐蚀。
2) 采用含时密度泛函方法TD-B3LYP方法研究了分子轴方向电偶极场对PuO3的激发态的影响,表明激发能与外电场的关系近似满足Grozema等提出的关系。由于激发能随电场增大而减小,表明在外电场作用下电子容易激发。
参考文献:
[1] LARSON D T, HASCHKE J M. XPS-AES characterization of plutonium oxides and oxide carbide. The existence of plutonium monoxide[J]. Inorg Chem, 1981, 20: 1945-1950.
[2] ALMEIDA T, COX L E, WARD J W. Gas adsorption studies on Pu metal by photoemission spectroscopy [J]. Surf Sci, 1993, 287: 141-145.
[3] STAKEBAKE J L, LARSON D T, HASCHKE J M. Characterization of the plutonium-water reaction Ⅱ: formation of a binary oxide containing Pu(Ⅵ) article [J]. J Alloys Compd, 1993, 202: 251-261.
[4] HASCHKE J M, RICHETTS T E. Adsorption of water on plutonium dioxide [J]. J Alloys Compd, 1997, 252: 148-156.
[5] HASCHKE J M, OVERSBY V M. Plutonium chemistry: a synthesis of experimental data and a quantitative model for plutonium oxide solubility [J]. J Nuclear Materials, 2002, 305: 187-201.
[6] MOREL J, BICKEL M, HILL C. Results of the international Pu-2000 exercise for plutonium isotopic composition measurements [J]. Apllied Radiation and Isotopes, 2004, 60: 607-614.
[7] ROUSSEL D S. Experimental values for241Am and239+240Pu Kd’s in French agricultural soils [J]. J Environmental Radioactivity, 2005, 79: 171-185.
[8] WANG H Y, ZHU Z H, JIANG G. Potential energy function for the ground state X3Σ-and excited state1Σ+of UO[J]. Molecular Phys, 2000, 13: 875-880.
[9] MENG D Q, ZHU Z H, JIANG G. Geometrical configuration of Pu4and the Jahn-teller effect[J]. J Molec Struct,2002, 610: 241-245.
[10] LI Q, ZHU Z H. Study of analytic potential energy function and stability for PuOn+with density functional theory[J]. J Molec Struct,2001, 10 (6): 501-505.
[11] LI Q, ZHU Z H, WANG H Y, et al. Potential energy function for PuO2+, PuH2+and PuN2+ions[J]. J Molec Struct:Theochem, 2002, 578: 177-180.
[12] 高涛, 王红艳, 易有根, 等. PuO分子X5Σ-的势能函数及热力学函数的量子力学计算[J]. 物理学报, 1999, 12: 2222-2227.
[13] 蒙大桥, 蒋 刚, 朱正和. Pu3体系的结构与势能函数[J]. 物理学报, 2001, 50: 1268-1273.
[14] JOHN M H, THOMAS H A. Plutonium hydride, sesquioxide and monoxide monohydride: pyrophoricty and catalysis of plutonium corrosion [J]. J Alloys and Compounds, 2001, 320: 58-71.
[15] HAY P J, MARTIN R L. Theoretical studies of the structures and vibrational frequencies of actinide compounds using relativistic effective core potentials with Hartree-Fock and density functional methods: UF6, NpF6, and PuF6[J]. J Chem Phys,1998, 109: 3875-3881.
[16] GROZEMA F C, TELESCA R, JOUKMAN H T. Excited state polarizabilities of conjugated molecules calculated using time dependent density functional theory [J]. J Chem Phys,2001, 115: 10014-10021.
[17] KJEELLBERG P, ZHI H, TONU P. Bacteriochlorophyll in electric field [J].J Phys Chem B, 2003, 107: 13737-13742.
[18] STEFAN G. Density functional calculations with configuration interaction for the excited states of molecules [J]. Chem Phys Lett, 1996, 259: 128-137.
[19] 陈晓军, 罗顺忠, 蒋树斌, 等. 外场下二甲基硅酮双自由基的从头计算研究[J]. 原子与分子物理学报, 2004, 21: 203-209.
[20] CRAMER C J. Essentials of Computational Chemistry [M]. England: Wiley, 2002, 441.
[21] FRISCH M J, TRUCKS G W, SCHEGEL H B. Gaussian03, Revision B 03[R]. Pittsburgh P A: Gaussian Inc, 2003.
[22] 刘华蓉, 葛学武, 倪永红, 等. 水溶液中金属团簇的脉冲辐解研究进展[J]. 化学物理学报, 2001, 14: 1-18.
猜你喜欢
中学生数理化(高中版.高考理化)(2021年11期)2022-01-18
中学生数理化(高中版.高考理化)(2021年3期)2021-05-21
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
科技视界(2019年27期)2019-11-05
中国交通信息化(2019年5期)2019-08-30
中国交通信息化(2017年1期)2017-06-08
中国交通信息化(2017年4期)2017-06-06
新高考·高一物理(2016年7期)2017-01-23
中国交通信息化(2017年10期)2017-01-14
