
用“点击化学”法合成新型他克林-四氢异喹啉异二联体*
2011-11-27 02:00郭永彪刘海波
合成化学 2011年3期
郭永彪, 刘海波, 许 明
(防化研究院,北京 102205)
他克林(Chart 1)是美国FDA批准的第一个用于治疗阿尔茨海默症的药物。他克林虽然作用明显,但肝毒性较大,尤其会引起转氨酶水平升高,从而限制了其临床应用,这促使人们不断对其进行结构改造,以期获得治疗效果好、副作用低的候选药物。近年来,人们以他克林作为与乙酰胆碱酯酶的外周位点或中心位点结合的片断,合成了大量具有双重作用位点的乙酰胆碱酯酶抑制剂[1~3],双他克林是其中的代表性化合物。
Sharpless课题组[4~6]在研究靶向诱导合成时,将“点击化学”[7]引入乙酰胆碱酯酶抑制剂的合成中,发现syn-TZ2/PA6,syn-TZ2PIQ-A6和syn-TZ2PIQ-A5(Chart 1)的抑制活性达到10-15mol·L-1。Bourne等[8]通过syn-TZ2/PA6与胆碱酯酶复合物的晶体结构证明,syn-TZ2/PA6不仅与乙酰胆碱酯酶的外周位点和中心位点结合,而且连接链中的三唑环与乙酰胆碱酯酶芳香峡谷中部的氨基酸形成氢键并相互堆垛。这说明三唑环不仅起到连接构建基团的作用,而且是一个大大促进与蛋白质结合的药效团。
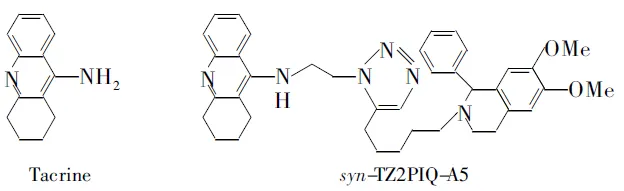
Chart1
Scheme1
本文拟通过“点击化学”法将三唑环引入他克林二联体中,以期得到具有三作用位点的乙酰胆碱酯酶抑制剂,从而提高抑制活性和选择性。基于此,本文设计并合成了3个他克林-四氢异喹啉异二联体(1a~1c, Scheme 1),其结构经NMR和HR-MS表征。其中1b和1c未见文献报道。合成路线如Scheme 1所示:从2-氯乙胺盐酸盐出发,经叠氮化与缩合反应制得9-(叠氮基乙基氨基)-1,2,3,4-四氢吖啶类衍生物(4a~4c); 以6-庚炔-1-醇为原料,利用微波反应合成了2-(6-庚炔基)-6,7-二甲氧基-1-苯基-1,2,3,4-四氢异喹啉(7);4与7通过“点击化学”法合成了1。
1 实验部分
1.1 仪器与试剂
XT4 100B型显微熔点仪(温度未经校正);VarianVMS 600 M型核磁共振仪(CDCl3为溶剂,TMS为内标);TSQ 7000型质谱仪;Varian CP-3800型气相色谱仪(GC); LC/MSDTOF液相色谱-质谱联用仪(LC-MS); CEM DISCOVER型微波反应仪。
2-氯乙胺盐酸盐[9],3[10]和6[11]按文献方法合成;硅胶200目~300目,青岛海洋化工厂;其余所用试剂为市售化学纯或分析纯。
1.2 合成
(1) 2-叠氮乙胺(2)的合成
在反应瓶中加水50 mL, 2-氯乙胺盐酸盐5.8 g(50 mmol)及叠氮化钠6.5 g(100 mmol),搅拌于80 ℃反应12 h。冷却至室温,用20%NaOH溶液调至pH≈10,用乙醚(3×50 mL)萃取,无水Na2SO4干燥,分馏收集83 ℃~85 ℃馏分得无色油状液体2,产率89.3%。
(2)4的合成(以4a为例)
在反应瓶中加入苯酚3.76 g(40 mmol), 9-氯-1,2,3,4-四氢吖啶(3a) 2.18 g(10 mmol),22.58 g(30 mmol)和碘化钠0.15 g(1 mmol),搅拌下缓慢升温至100 ℃,反应3 h。冷却至室温,加CH2Cl250 mL稀释,依次用10%NaOH溶液(40 mL),水(40 mL),饱和NaCl溶液(40 mL)洗涤,无水Na2SO4干燥,浓缩后经硅胶柱层析[洗脱剂: A=V(石油醚) ∶V(乙酸乙酯)=1 ∶1]分离得深黄色固体9-(叠氮基乙基氨基)-1,2,3,4-四氢吖啶(4a),产率95.5%, m.p.44 ℃~46 ℃。
用类似的方法合成浅黄色固体4b(产率98.7%, m.p.62 ℃~63 ℃)和4c(产率97.6%, m.p.63 ℃~64 ℃)。
4a~4c表征数据[14]与Scheme 1预期吻合。
(3) 6-庚炔-1-碘(5)的合成
在反应瓶中加入POCl318.2 mL(200 mmol),冰浴冷却,搅拌下缓慢滴加6-庚炔-1-醇5.6 g(50 mmol,保持内温低于10 ℃),滴毕,缓慢升温至室温反应至终点(GC监测)。缓慢倒入盛有200 g碎冰的大烧杯中,搅拌至碎冰完全融化,静置1 h,待烧杯底部有深红色油状物出现时,用二氯甲烷(2×50 mL)萃取,合并萃取液,用无水Na2SO4干燥,旋蒸脱溶,残余物减压分馏,收集68 ℃~70 ℃馏分得无色油状液体6-庚炔-1-氯。
在微波反应管(30 mL)中加入丙酮10 mL, 6-庚炔-1-氯1.96 g(15 mmol)和碘化钠4.50 g(30 mmol),置于微波反应器(40 W辐射)中,中速搅拌下于80 ℃反应1 h(GC监测)。冷却至室温,抽滤,滤液旋除绝大部分溶剂后加正己烷50 mL,过滤,滤液旋蒸至干得浅黄色油状液体5,产率90.0%。
(4)7的合成
在微波反应管(30 mL)中加入乙腈10 mL,61.35 g(5 mmol)及51.11 g(5 mmol),置于微波反应器(40 W辐射)中,中速搅拌下于80 ℃反应10 min(GC监测)。浓缩后经硅胶柱层析(洗脱剂:A=1 ∶2)分离得白色粉末7,产率93.6%, m.p.55 ℃~56 ℃; HR-MSm/z: Calcd for [M+H]+364.227 1, found 364.227 6。NMR表征数据[14]与Scheme 1预期吻合。

表 1 1a~1c的NMR数据
(5)1的合成(以1a为例)
在反应瓶中加入50%丁醇2 mL,4a75 mg (0.25 mmol),791 mg(0.25 mmol),五水硫酸铜6 mg(0.025 mmol)及抗坏血酸钠(NaAsc)10 mg(0.05 mmol),搅拌下于室温反应5 h(TLC跟踪)。加二氯甲烷20 mL稀释,依次用水(10 mL)和饱和NaCl溶液(10 mL)洗涤,无水Na2SO4干燥,浓缩后经硅胶柱层析(洗脱剂:A=1 ∶4)分离得浅黄色固体1a。用类似的方法合成浅黄色固体1b和深黄色固体1c, NMR数据见表1。
1a: 产率88.9%, m.p.64 ℃~67 ℃; HR-MSm/z: Calcd for [M+H]+631.375 5, found 631.372 7。
1b: 产率89.2%, m.p.103 ℃~105 ℃; HR-MSm/z: Calcd for [M+H]+665.336 5, found 665.336 4。
1c: 产率85.2%, m.p.65 ℃~67 ℃; HR-MSm/z: Calcd for [M+H]+665.336 5, found 665.335 3。
2 结果与讨论
2.1 合成4的方法改进
Sharpless等[4]首次合成了4a,其反应路线存在的问题是:(1) 中间体及目标化合物均需要柱分离纯化,效率低; (2)反应时间长、收率较低;(3)4a衍生物的合成效率低。
本文设计了新的合成路线(Scheme 1),以2-氯乙胺盐酸盐为原料经叠氮化得到2;2与3缩合得4。与文献方法比较,改进方法减少了柱分离纯化次数(两次[4]),简化了操作,提高了效率;缩短了反应时间(25 h[4]),明显提高了收率(73%[4])。尤其在合成4a衍生物4b和4c时充分体现了改进方法的优越性。
2.2 微波方法合成5和7
本文将微波合成技术引入5和7的合成中,实验结果与常规方法的比较见表2。从表2可以看出,5的合成采用常规与微波两种方法都可得到较高产率,但微波反应仅需要1 h;相比于常规条件下的48 h大大缩短了反应时间。采用微波法合成7,不仅大大缩短了反应时间,并且显著提高产率。

表 2 微波方法合成5和7与常规方法的比较
2.3 “点击化学”合成1
炔烃与叠氮发生Huisgen[3+2]环加成反应生成三唑是“点击化学”思想的重要体现。近年来对Cu(Ⅰ)催化[12,13]炔烃与叠氮反应得到选择性较高的1,4-二取代三唑化合物的研究较多,其反应机理为:末端炔先与Cu(Ⅰ)生成铜盐,然后铜和有机叠氮络合,再进一步关环得到六元环过渡态,通过重排得到三唑环的铜盐,氢化脱去Cu(Ⅰ)就得到1,4-二取代-1,2,3-三唑化合物。
Cu(Ⅰ)可来源于铜盐,如CuCl, CuBr, CuI等,但反应过程中需要隔绝空气;还可来源于Cu(0),反应过程不需隔绝空气,后处理简单,但反应时间较长;另外通过还原Cu(Ⅱ)得到,反应过程不需隔绝空气,反应时间较短。综合考虑,本文选择通过NaAsc还原五水硫酸铜原位制备Cu(Ⅰ)催化4与7反应生成1。
3 结论
本文设计新路线合成了9-(叠氮基乙基氨基)-1,2,3,4-四氢吖啶及其衍生物(4a~4c)。该方法具有反应时间短,后处理简单、收率高等优点,为今后合成基于他克林的三作用位点乙酰胆碱酯酶抑制剂提供重要中间体奠定了良好基础。
将微波合成技术引入碘代及亲核取代反应,大大缩短了反应时间,并可提高产率。
通过“点击化学”法,采用Cu(Ⅰ)催化成功地合成了三个他克林-四氢异喹啉异二联体(1a~1c),但均为1,4-二取代三唑化合物,1,5-二取代三唑化合物的合成无疑具有更大的挑战性,这些是我们今后研究的重点。
[1] Camps P, Formosa X, Galdeano C,etal. Pyrano[3,2-c]quinoline-6-chlorotacrine hybrids as a novel family of acetylcholinesterase andβ-amyloid-directed anti-alzheimer compounds[J].J Med Chem,2009,52(17):5367-5379.
[2] Camps P, Formosa X, Galdeano C,etal. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-alzheimer drug candidates[J].Chemico-Biological Interactions,2010,187:411-415.
[3] Jos H M Lange, Hein K A C Coolen, Martina A W van der Neut,etal. Design,synthesis,biological properties,and molecular modeling investigations of novel tacrine derivatives with a combination of acetylcholinesterase inhibition and cannabinoid CB1receptor antagonism[J].J Med Chem,2010,53(3):1338-1346.
[4] Lewis W G, Green L G, Grynszpan F,etal. Click chemistry in situ:Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks[J].Angew Chem Int Ed,2002,41(6):1053-1057.
[5] Manetsch R, Krasinski A, Radic Z,etal. In situ click chemistry:Enzyme inhibitors made to their own specifications[J].J Am Chem Soc,2004,126:12809-12818.
[6] Krasinski A, Radic Z, Manetsch R,etal. In situ selection of lead compounds by click chemistry:Target-guided optimization of acetylcholinesterase inhibitors[J].J Am Chem Soc,2005,127:6686-6692.
[7] Kolb H C, Finn M G, Sharpless K B. Click chemistry:Diverse chemical function from a few good reactions[J].Angew Chem Int Ed,2001,40:2004-2021.
[8] Bourne Y, Kolb H C, Radic Z,etal. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation[J].Natl Acad Sci,2004,101:1449-1454.
[9] Wolfgang S, Wolf M, Guenther C. Preparation of 2-chloroethylamine hydrochloride[P].DE 4 411 917 A1,1995.
[10] Recanatini M, Cavalli A, Belluti F,etal. SAR of 9-amino-1,2,3,4-tetrahydroacridine based acetylcholinesterase inhibitors:Synthesis,enzyme inhibitory activity,QSAR,and structure based CoMFA of tacrine analogues[J].J Med Chem,2000,43:2007-2018.
[11] Paul R, Coppola J A, Cohen E. 1-Phenyl-zphenethyl-1,2,3,4-tetrahydroisoquinolines.A new series of nonsteroidal female antifertility agents[J].J Med Chem,1972,15(7):720-726.
[12] Jawalekar A M, Meeuwenoord N, Overkleeft H S,etal. Conjugation of nucleosides and oligonucleotides by [3+2] cycloaddition[J]. J Org Chem,2008,73:287-290.
[13] Kalisiak J, Sharpless K B, Fokin V V. Efficient synthesis of 2-substituted-1,2,3-triazoles[J].Org Lett,2008,10(15):3171-3174.
[14]4a:1H NMRδ: 7.93~7.91(m, 2H), 7.58~7.54(m, 1H), 7.40~7.27(m, 1H), 4.21(s, 1H), 3.57(t,J=5.1 Hz, 2H), 3.50(t,J=5.4 Hz, 2H), 3.08(t,J=6.0 Hz, 2H), 2.77(t,J=5.7 Hz, 2H), 1.96~1.91(m, 4H);13C NMRδ: 158.92, 147.30, 147.41, 128.99, 128.35, 124.24, 122.24, 120.85, 118.20, 52.09, 47.78, 34.05, 24.71, 22.95, 22.74; HR-MSm/z: Calcd for [M+H]+268.155 7, found 268.158 2.4b:1H NMRδ: 7.92(d,J=9.0 Hz, 1H), 7.41(d,J=2.4 Hz, 1H), 7.30~7.28(m, 2H), 3.91(t,J=5.4 Hz, 1H), 3.58(t,J=5.4 Hz, 1H), 2.79(t,J=5.7 Hz, 1H), 2.46(t,J=5.7 Hz, 1H), 1.79~1.75(m, 4H);13C NMRδ: 156.66, 151.80, 138.66, 138.35, 126.69, 125.90, 118.98, 114.12, 113.37, 50.89, 46.96, 28.21, 23.48, 21.40, 20.30; HR-MSm/z: Calcd for [M+H]+302.116 7, found 302.117 1.4c: m.p.63 ℃~64 ℃;1H NMRδ: 7.85~7.84(m, 1H), 7.44~7.40(m, 2H), 5.79(t,J=6.9 Hz, 1H), 3.53(t,J=5.7 Hz, 2H), 3.38~3.35(m, 2H), 3.07(t,J=6.9 Hz, 2H), 2.80(t,J=6.9 Hz, 2H), 1.98~1.84(m, 4H);13C NMRδ: 159.86, 150.46, 144.88, 129.01, 127.80, 127.65, 127.42, 121.98, 119.06, 51.86, 47.74, 33.50, 26.39, 22.98, 22.68; HR-MSm/z: Calcd for [M+H]+302.116 7, found 302.118 8.7:1H NMRδ: 7.54~7.28(m, 5H), 6.69(s, 1H), 6.13(s, 1H), 5.28(s, 1H), 3.89(s, 3H), 3.64(s, 3H), 3.33~3.26(m, 1H), 3.05~3.00(m, 2H), 2.62~2.47(m, 2H), 2.36(m, 1H), 2.19 (m, 2H), 1.76(s, 1H), 1.54~1.24(m, 6H);13C NMRδ: 149.40, 146.89, 144.24, 130.11, 129.48, 128.01, 126.97, 126.87, 111.58, 110.68, 83.67, 68.78, 66.96, 55.97, 55.89, 53.31, 46.38, 27.51, 25.80, 24.11, 22.67, 18.10.
猜你喜欢
商品与质量(2019年32期)2019-11-29
儿童故事画报·发现号趣味百科(2019年5期)2019-07-14
火工品(2018年1期)2018-05-03
中国资源综合利用(2017年3期)2018-01-22
中国果菜(2016年9期)2016-03-01
合成化学(2015年9期)2016-01-17
儿童故事画报·发现号趣味百科(2015年6期)2015-08-17
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
当代医学(2014年36期)2014-07-31
火炸药学报(2014年5期)2014-03-20
