
宏基因组技术研究进展
2012-01-24 02:39印蕾高向东顾觉奋
中国医药生物技术 2012年3期
印蕾,高向东,顾觉奋
微生物支撑着地球上的物质循环和生命延续,除了自身可作为新基因资源的重要来源外,还可产生对人类有价值的活性物质。然而,传统纯培养方法严重限制了人们对微生物资源的认识和开发。一方面,随着对微生物活性产物研究的深入,微生物往往被重复培养和筛选,从传统方法来筛选新活性物质的几率不断下降。另一方面,多达 99% 的微生物在现有实验条件和技术下尚未得到纯培养,其中蕴含着大量潜能微生物和基因资源[1]。近年来,随着基因组学在各个领域的渗入和现代分子技术的逐渐成熟,宏基因组学(Metagenomics)应运而生,开启了环境微生物研究的新方向。1986年,Olsen 等[2]提出直接从环境中克隆核糖体小亚基 DNA(16SrDNA),首次运用非纯培养的分子生物学方法研究展开微生物多样性研究。随着环境基因组学(Environmental genomics)概念的出现[3]以及第一个海洋微生物宏基因组文库的建立[4],宏基因组学研究开始受到广泛关注。1998 年,Handelsman 等[5]在前人研究的基础上正式提出了宏基因组(Metagenone)的概念,即“特定生态环境中所有生物遗传物质的总和”。宏基因组学则以环境样品中微生物群体基因组为研究对象,采用功能基因筛选和测序分析等研究工具,从不可培养微生物中来寻找新基因或开发新生物活性物质[6]。宏基因组学的产生使人们摆脱了物种界限,克服了传统微生物培养方式的缺陷,扩大了微生物资源的利用,掀开了环境微生物研究的新篇章。
1 宏基因组学研究策略
区别于传统培养途径(图 1)[7],宏基因组学的研究过程包括从环境样品中提取基因组 DNA;载体连接;转化宿主细胞,形成一个重组的 DNA 文库;筛选目的克隆 4 个步骤。
1.1 提取 DNA
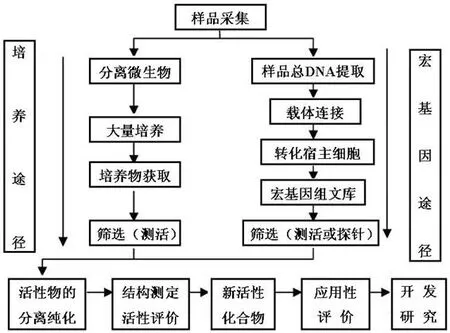
图 1 传统培养方法和宏基因组筛选微生物活性产物研究流程的简略比较
宏基因组文库构建的第一步是要得到高质量的 DNA,既应尽力保证环境样品中总 DNA 的完全提取,又要得到较大的片段以获取表达所需的完整的目的基因或基因簇。所以 DNA 提取时,应尽量在最大提取量和最小剪切力间保持平衡。目前已有许多商品化的宏基因组 DNA 提取试剂盒可用。最常用的两种提取方法为直接裂解法和间接提取法。前者又称原位裂解法,是将环境样品直接悬浮在裂解缓冲液中处理后再抽提纯化,可分为物理法(如高温或冻融法、液氮研磨法、超声法、玻璃珠击打法等)、化学法、酶法(如蛋白酶 K)。此法所得到的 DNA 能更好地代表样品微生物的多样性,且快速有效、成本低、DNA 提取率高、重复性好。但由于难以完全去除样品中的酚类物质,且存在强烈的机械剪切作用,导致所提取的 DNA 片段较小(1 ~ 50 kb),常用于构建小片段基因组文库。间接提取法又称异位裂解法,先用物理方法(如尼可登介质密度梯度离心法)将微生物细胞从环境样品中分离出来,然后用较温和的方法抽提DNA。此法可获得纯度较高的大片段 DNA(20 ~ 500 kb),适用于构建大片段插入文库,但操作繁琐,成本高,分离过程中容易丢失物种信息,温和条件下一些细胞壁较厚的微生物基因组也不容易抽提出来。需要注意的是,环境样品中的诸多因素如酚类化合物及高浓度的金属离子会干扰提取及下游基因工程操作中工具酶的活性,应尽量排除。此外,土壤中含有的腐植酸类物质可以用聚乙烯吡咯烷酮(PVP)或漆酶来处理。用于 RNA 提取的土壤样品可以加入RNAlater避免 RNA 的降解[8]。
为了更好地反映环境中的微生物种群,在克隆之前可用不同方法对目的基因或基因组进行富集,通过提高样品基因组 DNA 的特异性以利于进一步分离筛选。例如对富含 GC碱基基因的富集,可以直接从环境中提取 DNA,然后利用超速离心富集不同 GC 比例的 DNA。此外,还可运用稳定同位素示踪技术,利用一些比较稳定的同位素如13C、18O、15N 标记底物并结合密度梯度离心技术来分离大分子 DNA和 RNA。而在采集一些低营养环境(如极地环境或岩石)样品无法获得足够克隆的 DNA 时,则可运用兴新的多重置换扩增技术(multiple displacement amplification,MDA)从少量细胞中获得更多遗传信息。
1.2 载体选择
宏基因组文库的构建需要适宜的克隆载体,主要从利于目的基因扩增以及易于控制活性物质表达量等方面选择合适的载体。按照不同功能,常用的载体大致可分为以下3 类。
⑴克隆载体:以扩增 DNA 为目的、不需要得到目的基因编码蛋白的载体。早期构建宏基因组文库以质粒载体为主,操作简便,拷贝数高,但插入片段小于 10 kb,不适用于大基因簇筛选。产生微生物次级代谢产物的代谢途径由多基因簇调控,应该尽量插入大片段 DNA 以获得完整的代谢途径多基因簇。对于这些含较大基因簇或大片段 DNA的样品,多采用 Cosimid 或 Fosmid载体(35 ~ 45 kb),前者比较常用,而后者插入稳定性高;此外还有容量更大的细菌人工染色体(BAC)载体(通常为 100 kb),但克隆效率较低。
⑵表达载体:能使外源目的基因在宿主细胞中转录和表达的功能性载体。为了提高宏基因的表达,便于重组克隆的筛选和活性检测,可以直接利用表达载体构建宏基因组文库,但可插入的宏基因片段一般小于 10 kb,只适合于筛选单一基因或小片段的操纵子产物。
⑶穿梭载体:含有两个亲缘关系不同的复制子,能在不同宿主菌如原核大肠杆菌和真核酵母菌中复制。利用穿梭载体可扩大宿主范围,将不同文库中的信息进行转移,促使并提高外源基因的表达。
1.3 宿主菌株选择
宿主细胞的选择主要应考虑转化稳定性、转化效率、宏基因的表达量、目标性状等。研究目的不同,选择的宿主菌株也有所差异。常用的宿主菌有:
⑴大肠杆菌:大肠杆菌(Escherichia coli)因其具有操作简单、繁殖迅速、培养代谢易于控制等优点[9],是一种常用的宿主细胞。但是由于环境样品总 DNA 中占很大比例的真核基因组 DNA 在细菌宿主中不能表达,因此这部分基因的筛选受到了极大的限制。
⑵链霉菌属:链霉菌(Streptomyces spp.)具备天然的抗生素生产机制,其编码次生代谢产物生物合成的基因成簇分布,而这些基因的表达同时受细胞内和细胞外信号转导的调控。在链霉菌中已经表达出一些杂合抗生素、非核糖体多肽和一些聚酮,是目前次级代谢产物异源表达最合适的宿主菌。
⑶恶臭假单胞菌:恶臭假单胞菌(Pseudomonas putida)也是次生代谢产物的产生者。通过基因重组,恶臭假单胞菌可以获得链霉菌噬菌体的插入位点,接受链霉菌 BAC 穿梭载体克隆并且能够稳定地表达[10]。
1.4 文库筛选
宏基因组文库筛选主要可分为以下 4 种。其中,基于目的克隆功能和基于核酸序列差异的筛选是两种最常见的策略:
⑴功能驱动筛选:功能驱动筛选(function driven screening)是根据重组克隆具有的生物活性进行筛选的方法,常见于工农业和医药业中酶类蛋白及抗生素等全新天然产物的发现。具体策略包括:①在含有化学染料和不可溶或发光的酶反应底物的培养基中,对具有产生特殊活性功能的克隆根据不同表型特征进行直接筛选。②利用含外源基因的宿主菌株与其突变体在选择性条件下功能互补生长的特性在培养基上筛选阳性克隆。功能驱动筛选虽然不需了解基因序列信息,但必须依赖目的基因在宿主菌中的有效表达,因而要求选择合适的宿主菌株,同时应克隆到完整的基因或基因簇。一旦基因的某个组件在克隆过程中被破坏将使基因无法表达,也就不能根据表型加以筛选。Park 等[11]还发现,宏基因组中不确定阅读框(URFs)与宿主菌(如 E.coli)的开放阅读框架(ORF)序列存在的大量差异也是使该法筛选阳性克隆率低的原因。此方法可筛选到全新的天然产物和蛋白基因,但工作量大、效率低,检测手段有一定局限。
⑵序列驱动筛选:序列驱动筛选(sequence driven screening)基于已知的基因或基因表达产物的保守序列设计探针或 PCR 引物,通过 PCR 扩增或核酸杂交筛选目的克隆。序列驱动筛选适用于系统发育学中标志基因如 16sRNA基因的鉴定或高度保守的酶基因如聚酮合成酶的发现。其优点是不依赖目的基因在宿主菌株中的表达,但由于探针的设计必须建立在已知基因序列的基础上,因而无法筛选出与现有基因序列完全不同的新基因。序列驱动筛选通常需要基因芯片等高通量测序技术和生物信息学分析软件的支持[12],相对功能筛选成本更高。
⑶化合物结构水平的筛选:化合物结构水平的筛选(screening on compound structure)是基于不同物质在色谱分析中产生不同吸收峰的原理,通过比较转入外源基因前后宿主细胞或表达产物的色谱图可迅速筛选出能生成新结构化合物的克隆子,再结合生物学手段进一步确定活性。此方法效率低、费用高、工作量大。
⑷底物诱导基因表达筛选:底物诱导基因表达筛选(substrate-induced gene expression screening,SIGEX)是利用合适的底物进行诱导,使目的基因表达进行筛选的方法,并可从底物推断出未知基因的功能,尤其适用于在工业上筛选分解代谢相关基因和酶基因。克隆 SIGEX 法可用于高通量筛选,而且不需要对底物进行修饰,并可从底物直接推断出未知基因的功能。该法的缺点:①对目标基因的结构性和宿主适应性很敏感;②用于诱导的底物必须要进入细胞质,否则无法诱导目的基因的表达;③本可诱导表达的基因由于宿主的改变会无法被诱导表达;④对于无需诱导即可表达或同时受多因素诱导的基因不适用。
2 在医药领域的研究进展
由于微生物天然产物开发技术陈旧,越来越多的已知结构化合物被重复发现,新的微生物来源天然药物的开发也变得越发困难。但是宏基因组技术突破了纯培养的界限,无需特定的微生物来源,成为目前药物开发的新方向[13]。
2.1 环境微生物多样性研究
近年来,宏基因组技术已逐渐成为继 16SrDNA 克隆之后研究微生物系统发育和功能的又一有力工具,研究获得的大量基因信息帮助我们对生物种群的结构与进化关系,以及不同生物群落中微生物的多样性有了新的认识[14]。2000年,Rondon 等[15]利用土壤环境,选用 BAC 载体构建了两个大片段 DNA 插入宏基因组文库(SL1 和 SL2),共获得了 1.1 Gb 的 DNA 分子,通过对 SL1 文库 16SrRNA 基因序列分析发现了低 GC 比例的革兰阳性菌、Acidobacterium、Cytophagales 和 Proteobacteria 等微生物类群。Venter 等[16]以序列筛选分析为基础,对马尾藻海域表面海水的宏基因组进行了 1.6 Gb DNA 序列测定,利用生物信息学软件分析后进行基因组组装,鉴定出大量新基因,发现了 148 种新微生物和 782 种光受体。
2.2 已发现的抗生素及其相关基因及抗生素抗性研究
加拿大 TerraGen Discovery 公司最先运用穿梭 Cosmid载体,在以链霉菌为宿主的宏基因组文库中从化合物结构水平筛选到了具有抗菌活性的 5 种全新的小分子物质Terragine A、B、C、D、E(图 2)。Kosan Technology 公司从宏基因组中发现了阿霉素、红霉素、四环素、Rapamycin 和FK506 等一系列天然产物。2002 年,Gillespie 等[17]在构建的土壤宏基因组文库中,筛选获得两种具有广谱抗菌活性的物质 Turbomycin A 和 B(图 2)。Banik 和 Brady[18]根据在万古霉素和替考拉宁样糖肽基因簇中发现的保守的氧化偶联酶 OxyC 设计引物从土壤宏基因组文库中筛选得到一系列糖肽合成编码基因簇,通过重组产生了 15 个新的阴离子(硫酸)糖肽类抗生素。从多个海洋宏基因组中发现的几百个新聚酮类化合物生物合成途径对聚酮化合物的结构改造以及开发更多具有抗菌和抗肿瘤活性的大环内酯类抗生素有着重大作用[19]。
有研究表明,抗生素抗性基因(ARGs)可随着各种排泄物排放到各种环境中,包括河流沉积物、灌溉渠、牛奶加工厂的出水、污水处理池等,并可通过水平转移到其他生物中,造成二次污染,因而应用宏基因组学也可以直接从环境样品中获得未培养细菌的抗性基因,用于耐药性研究。2003年,Diaz-Torres 等[20]通过对人体唾液宏基因组文库的筛选得到一种新的对四环素具有很好抗性的基因 Tet(37)。2008年,Mori 等[21]通过活性污泥宏基因组文库筛选获得两种不同的博来霉素抗性基因。van Elsas 等[22]从具有抗药性的土壤中筛选出几个 40 kb 的具有潜在抗性的基因。
2.3 酶类物质的发现
生物催化剂也是宏基因组学研究的重点方向。目前已构建了土壤、海水、污泥、人体口腔和胃肠道等宏基因组文库,从中筛选到具有不同催化活力的蛋白酶、脂肪酶、聚糖酶、氧化酶、核酸酶、生物素合成酶系等(表 1)。
2.4 病毒及人体宏基因组学研究

图 2 化合物 Turbomycin A、B 和 Terragine A、B、C、D、E 结构式
过去十年,病毒宏基因组学研究的开展帮助人们从多样的环境中快速有效地发现了许多之前未知的新病毒及其基因,成为传染病预防和诊断的新武器[23]。2009 年建立的人体微生物工程基因计划(human microbiome project)[24]旨在描绘肠道、口腔、皮肤等人体中的微生物群,其规模和广度将远远超过人类基因组计划。据估计,人类宏基因组计划将发现超过 100 万个新基因,这对于研究疾病发生机制、控制药物毒性、开发新药等都有着积极的意义。
在我国利用宏基因组技术开发药物的案例较少,在“十五”期间,对东太平洋深海沉积物进行了采集研究,利用基因大片段提取技术,构建了柯斯质粒等大分子文库和片段大小为 6 ~ 23 kb的各类质粒、噬菌体库,筛选获得 7 个具有较好抗肿瘤活性的宏基因组克隆子,还发现了一些新的来源于未培养微生物的功能基因。
3 展望与挑战
迄今为止,宏基因组技术是最有效的开发和利用未培养微生物资源的工具,已有超过 210 种不同宏基因组被测序,这些基因组来自于不同的环境,例如传统的土壤资源、海洋资源、一些极端环境、人类肠道和粪便等[25]。随着基因组测序、基因工程、蛋白质工程和代谢工程等生物技术的兴起,宏基因组学也迎来了更多的挑战与机遇。
宏基因文库存储的仅仅是微生物的基因信息,而并非菌体本身,这一点极大地限制了对微生物遗传背景的了解和对基因表达的有效调控,可能导致外源宏基因在宿主细胞中不表达或低表达,进而丢失许多环境微生物的天然产物。此外,宏基因组文库构建中样本 DNA 的提取不完整,载体和宿主菌的有限性,以及高通量筛选技术的局限性都是今后宏基因技术着力要解决的缺陷。

表 1 从宏基因组中已发现的酶类物质
令人欣喜的是,第二代测序技术的出现和大规模 DNA测序平台的广泛使用大大缩减了 DNA 的测序成本,将极大地促进宏基因组学的发展。例如,有效的生物信息学计算方法、宏基因组数据分析和功能预测软件的开发与应用[26]对宏基因组学的发展都具有积极的影响。此外,与宏基因组的策略相同的宏转录组学和宏蛋白质组学的发展填补了宏基因组的某些不足[27]。宏基因组学提供环境中总 DNA 的信息,宏转录组学提供实时的环境基因表达信息,宏蛋白质组学可以提供表达产物的功能信息。把这些“组学”联系起来,有助于从基因到蛋白质的全面研究,极大地丰富人类对自然界的认识。有文章分析,将宏基因组与组合生物合成及纳米技术结合,对于开发微生物来源的天然活性产物会有强大的促进作用[28]。
在新兴技术的帮助下,面对 99% 以上不能获得纯培养的广大微生物资源,宏基因组学不仅是我们获得各种基因资源的一个有效手段,更是对于新药研发,人类健康及生态环境认识的一个很好的工具,它必将获得更多的重视。
[1] Daniel R. The metagenomic of soil. Nat Rev Microbiol, 2005, 3(6):470-478.
[2] Olsen GJ, Lane DJ, Giovannoni SJ, et al. Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol, 1986, 40:337-365.
[3] Schmidt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing.J Bacteriol, 1991, 173(14):4371-4378.
[4] Stein JL, Mar sh TL, Wu KY, et al. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol, 1996, 178(3):591-599.
[5] Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frotier for natural products. Chem Biol, 1998, 5(10):245-249.
[6] Rajendhran J, Gunasekaran P. Strategies for accessing soil metagenome for desired applications. Biotechnol Adv, 2008, 26(6):576-590.
[7] Yan B, Hong K, Xu Y, et al. Metagenome cloning — a new approach for novel microbial bioactive compounds discovery. Microbiology,2005, 32(1):113-117. (in Chinese)阎冰, 洪葵, 许云, 等. 宏基因组克隆——微生物活性物质筛选的新途径. 微生物学通报, 2005, 32(1):113-117.
[8] Grant S, Grant WD, Cowan DA, et al. Identification of eukaryotic open reading frames in metagenomic cDNA libraries made from environmental samples. Appl Environ Microbiol, 2006, 72(1):135-143.
[9] Taupp M, Mewis K, Hallam SJ. The art and design of functional metagenomic screens. Curr Opin Biotechnol, 2011, 22(3):465-472.
[10] Martinez A, Kolvek SJ, Yip CL, et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol,2004, 70(4):2452-2463.
[11] Park SH, Cheong DE, Lee JY, et al. Analyses of the structural organization of unidenti fi ed open reading frames from metagenome.Biochem Biophys Res Commun, 2007, 356(4):961-967.
[12] Scholz MB, Lo CC, Chain PS. Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr Opin Biotechnol, 2012, 23(1):9-15.
[13] Lefevre F, Robe P, Jarrin C, et al. Drugs from hidden bugs: their discovery via untapped resources. Res Microbiol, 2008, 159(3):153-161.
[14] McHardy AC, Rigoutsos I. What’s in the mix: phylogenetic classi fi cation of metagenome sequence samples? Curr Opin Microbiol,2007, 10(5):499-503.
[15] Rondon MR, August PR, Bettermann AD, et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microb, 2000,66(6):2541-2547.
[16] Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science, 2004, 304 (5667):66-74.
[17] Gillespie DE, Brady SF, Bettermann AD, et al. Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Appl Environ Microbiol, 2002, 68(9):4301-4306.
[18] Banik JJ, Brady SF. Cloning and characterization of new glycopeptide gene clusters found in an environmental DNA megalibrary. Proc Natl Acad Sci U S A, 2008, 105(45):17273-17277.
[19] Hochmuth T, Piel J. Polyketide synthases of bacterial symbionts in sponges--evolution-based applications in natural products research.Phytochemistry, 2009, 70(15-16):1841-1849.
[20] Diaz-Torres ML, McNab R, Spratt DA, et al. Novel tetracycline resistance determinant from the oral metagenome. Antimicrob Agents Chemother, 2003, 47(4):1430-1432.
[21] Mori T, Mizuta S, Suenaga H, et al. Metagenomic screening for bleomycin resistance genes. Appl Environ Microbiol, 2008, 74(21):6803-6805.
[22] van Elsas JD, Speksnijder AJ, van Overbeek LS. A procedure for the metagenomics exploration of disease-suppressive soils. J Microbiol Methods, 2008, 75(3):515-522.
[23] Mokili JL, Rohwer F, Dutilh BE. Metagenomics and future perspectives in virus discovery. Curr Opin Virol, 2012, 2(1):63-77.
[24] NIH HMP Working Group, Peterson J, Garges S, et al. The NIH human microbiome project. Genome Res, 2009, 19(12):2317-2323.
[25] Meyer F, Paarmann D, D'Souza M, et al. The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 2008,9:386.
[26] Wooley JC, Ye Y. Metagenomics: facts and artifacts, and computational challenges. J Comput Sci Technol, 2009, 25(1):71-81.
[27] Simon C, Daniel R. Metagenomic analyses: past and future trends.Appl Environ Microbiol, 2011, 77(4):1153-1161.
[28] Singh BK, Macdonald CA. Drug discovery from uncultivable microorganisms. Drug Discov Today, 2010, 15(78):792-799.
猜你喜欢
猪业科学(2021年3期)2021-05-21
今日农业(2020年19期)2020-12-14
科学(2020年3期)2020-11-26
幽默大师(2020年10期)2020-11-10
科学(2020年2期)2020-08-24
科学(2020年2期)2020-08-24
当代水产(2020年3期)2020-06-15
中华诗词(2019年1期)2019-11-14
猪业科学(2018年4期)2018-05-19
中华老年多器官疾病杂志(2016年9期)2016-04-28
