
TiO2纳米管修饰镍电极及其光催化辅助电解水制氢性能研究
2012-09-15 11:46何洪波陈爱平马董海军李春忠
无机化学学报 2012年10期
何洪波 常 明 陈爱平马 磊 董海军 李春忠
(华东理工大学材料科学与工程学院,超细材料制备与应用教育部重点实验室,上海 200237)
TiO2纳米管修饰镍电极及其光催化辅助电解水制氢性能研究
何洪波 常 明 陈爱平*马 磊 董海军 李春忠
(华东理工大学材料科学与工程学院,超细材料制备与应用教育部重点实验室,上海 200237)
本文通过水热法制备二氧化钛纳米管(TiO2NT),并用制备的TiO2NT对碱性电解水制氢装置的镍片阳极进行修饰,在电解水的基础上,通过光催化与电解过程的耦合,提出并实现了光催化辅助电解水制氢过程。通过XRD、UV-Vis、FE-SEM、AFM和光催化辅助电解水制氢等方法对试样的结构和性能进行了表征和测试。结果表明,在紫外光照条件下,用TiO2NT修饰镍片阳极的光催化辅助电解水过程的产氢速率比单纯电解水提高了61%。
TiO2纳米管;镍电极修饰;电解水;光催化;制氢
能源危机和环境污染是当今社会发展面临的严峻挑战。氢能因其高效和清洁被普遍推崇为最适当的能源载体。氢气是二次能源,在目前各种制氢技术中,电解水制氢工艺成熟,从环保和原料的角度被视为通向氢经济的较佳途径[1-4]。
1972年日本学者Fujishima和Honda[5]对光照二氧化钛电极导致水分解产生氢气这一现象的发现,揭示了利用太阳能裂解水制氢的可能性,其巨大的应用前景立即吸引了全世界众多研究者从事半导体光电化学制氢的研究[6-8]。现有的光电化学制氢研究,都是以光催化为基础,通过施加较低的直流偏压(一般都低于1 V,远低于电解水所需的理论电压1.23 V)构成电辅助光催化过程。虽然围绕光催化剂设计和筛选[9-14]、添加氢牺牲剂的电解质溶液的选择、采用真空抽气等特殊的制氢工艺、以及通过施加较低的直流偏压构成电辅助光催化过程 (光电化学制氢过程)等方面,进行了大量的工作,但以光催化为基础的分解水制氢过程的量子效率没有根本性的突破[15-16]。
将太阳能利用与电解水制氢结合在一起的工作,主要集中在如何把太阳能转化为电能的光伏技术与电解水制氢技术相结合方面。在已工业应用的碱性电解水制氢技术的改进研究中,主要集中在筛选金属基合金电极,达到降低电极的析氢析氧过电位、降低电耗等目的,但也没有取得显著的收效[17-18]。
TiO2纳米管由于具有更大的比表面积、更高的吸附能力和传导电子的能力,改善了颗粒TiO2的光电催化性能[19-22],受到了广泛的关注。Mor[23]等报道了TiO2纳米管阵列作阳极,Pt作阴极,1 V偏压,337 nm光照等条件下,光电转换效率IPCE超过90%。
本文改变目前光电化学制氢研究中以光催化为基础的思路,以成熟的碱性电解水制氢技术为基础,将TiO2纳米管光催化剂涂覆在电解池的镍片阳极上,通过光对阳极的辐照,借助纳米半导体光催化剂上产生的光生空穴所具有的强氧化活性,改善电解池阳极的析氧性能,在电解池直流电压的驱动下,光生电子及时迁移到阴极,促进阴极的析氢过程,将光催化与电解水制氢技术有机地耦合在一起,提出并实现了光催化辅助电解水制氢(WEAP)过程,提高了电解水的产氢速率。
1 实验部分
1.1 主要试剂与仪器
采用场发射扫描电镜(FE-SEM,日本JEOL公司JSM-6700F型)、透射电镜 (TEM,日本JEOL公司JEM-1230型)、原子力显微镜(AFM,NanoScope III a MultiMode,Veeco)观察TiO2纳米管薄膜的形貌;利用X射线衍射仪 (XRD,日本理学公司Rigaku D/Max 2550 VB/PC型)对二氧化钛粉体及二氧化钛纳米管的晶相进行分析;用紫外-可见吸收分光光度计(美国Varian公司Cary500 UV-Vis Spectro photometer)进行样品光吸收性能的测试。
试剂:Degussa 公司的 P25 TiO2(工业品),NaOH和浓盐酸均为分析纯试剂,购于国药集团化学试剂有限公司。
1.2 TiO2纳米管的制备
本实验以水热法[24-25]制备TiO2纳米管。称取0.5 g P25纳米粉体置于装有50 mL浓度为10 mol·L-1NaOH的内衬为聚四氟乙烯的高压釜中,密封后放在烘箱中383 K保温48 h后取出,自然冷却到室温,倒出溶液,用蒸馏水将沉淀物洗涤至中性后,在0.1 mol·L-1的稀盐酸中超声分散,使 Na+被 H+取代,继续用蒸馏水洗至中性,过滤后滤饼用于涂膜。
1.3 涂 膜
取一定量的洗至中性的TiO2纳米管滤饼,加入无水乙醇混合均匀,超声分散制成TiO2涂膜浆料。将一定量的TiO2浆料滴在Ni电极基片上,使用旋转涂膜机涂膜,待膜自然干燥后,于60℃下烘干,使膜结合更牢固。称量等质量的P25粉末用同样的方法在镍片上涂膜。
1.4 光催化辅助电解水制氢实验
在传统的碱性水电解池的基础上[20],设计的光催化辅助电解水制氢的实验装置如图1。
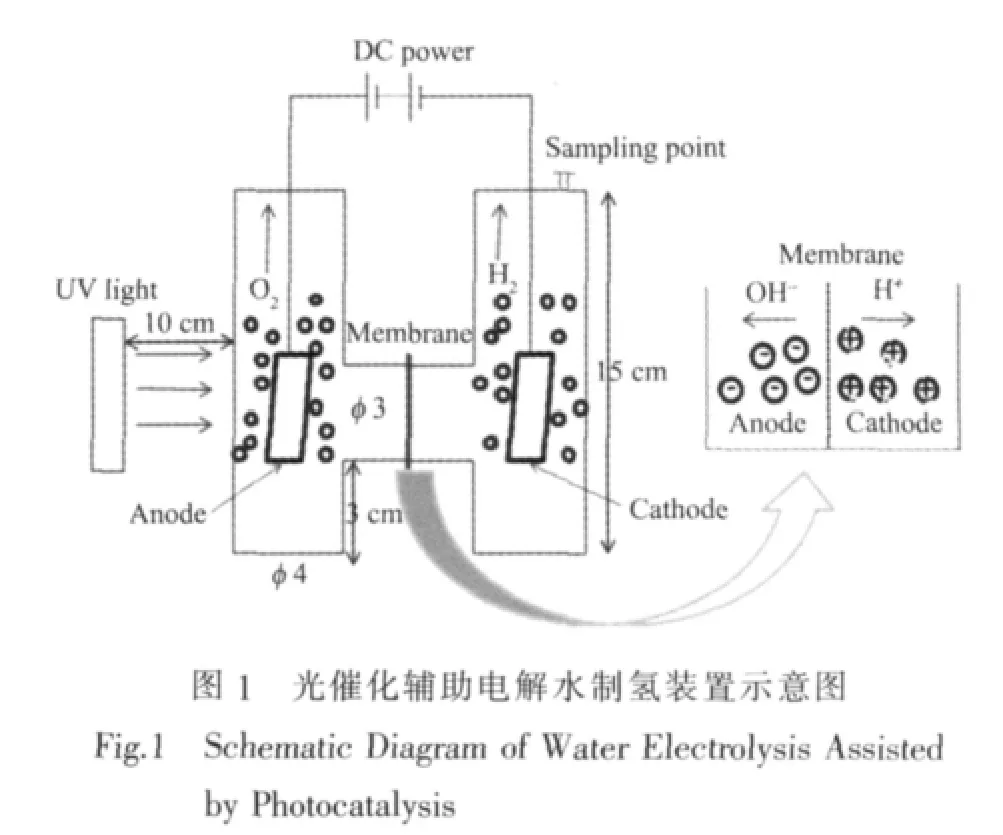
以工业化碱性水电解池为参考,采用的相关材料如下:阴极为 Ni-Cr 15×30×0.5 mm,阳极为 Ni 15×30×0.5 mm,全氟磺酸离子交换膜(φ 40 mm,厚 180 μm)作为阴极区与阳极区的隔膜,电解液为30%NaOH溶液。以TiO2光催化剂修饰的镍片为阳极,以紫外灯(150 W,主波长365 nm,上海亚明灯泡厂)为光源辐照阳极,构成光催化辅助电解水过程。产生的氢气量用气相色谱法测定。气相色谱仪以高纯氮气为载气,使用热导池(TCD)检测器。配制不同体积比的氢气标样,进样量为100 μL,得到氢气含量与峰面积的关系式:
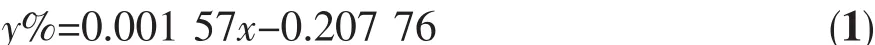
上式中y%:氢气体积百分含量,x:色谱峰峰面积,适用的氢气浓度范围为0.05%~80%。
由于阴极区最初全部是空气,当有H2生成时,阴极区的气体为H2含量不断增大的H2与空气的混合物,每隔一段时间用100 μL微量注射器从阴极区取样,注射到色谱仪中测其峰面积,用式(1)计算氢气含量,比较产氢速率。
3 结果与讨论
3.1 TiO2纳米管薄膜的形貌表征
图2a、2b分别为制备的TiO2纳米管滤饼的TEM和SEM图,可以看出P25纳米颗粒经水热反应后已全部转化成了TiO2纳米管,平均管外径约为10 nm,纳米管生长良好。从图2c中可以看出TiO2纳米管均匀地涂覆在Ni片上,管外径约为7~12 nm,与TiO2纳米管滤饼的形貌分析结果相当,说明涂膜过程不会改变TiO2纳米管结构。从图2d的膜层侧面观察可知,TiO2纳米管薄膜的厚度为6~7 μm。
图3是修饰在镍片上的TiO2纳米管薄膜的AFM表面形貌图,分析结果显示,薄膜表面致密,分布均匀,粗糙度(RMS)为 15.82 nm。


3.2 XRD分析
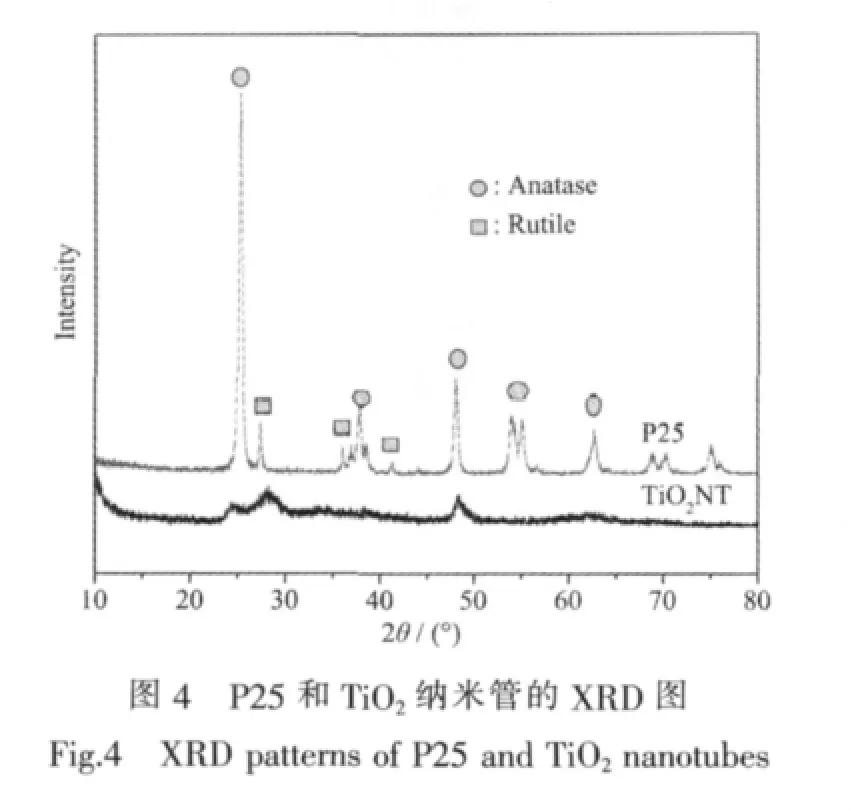
从图4所示的XRD图可以看出,原料P25是锐钛矿相和金红石相的混晶结构,锐钛矿与金红石的质量比约为80/20,而经过水热处理转变成TiO2纳米管后,锐钛矿与金红石的质量比变为50/50,并且衍射峰强度显著降低,说明结晶度显著下降。水热处理后生成的TiO2纳米管的衍射峰变宽,表明其晶粒变小。纳米级TiO2具有量子尺寸效应,颗粒变小,能隙变宽,导带电位更负,价带电位更正,将具有更强的氧化还原能力;对光生载流子被受体俘获过程的研究发现,粒径越小,光生电荷分离效率越高,则电子-空穴对复合几率越小,有利于TiO2光催化活性的提高[21]。
3.3 UV-Vis分析
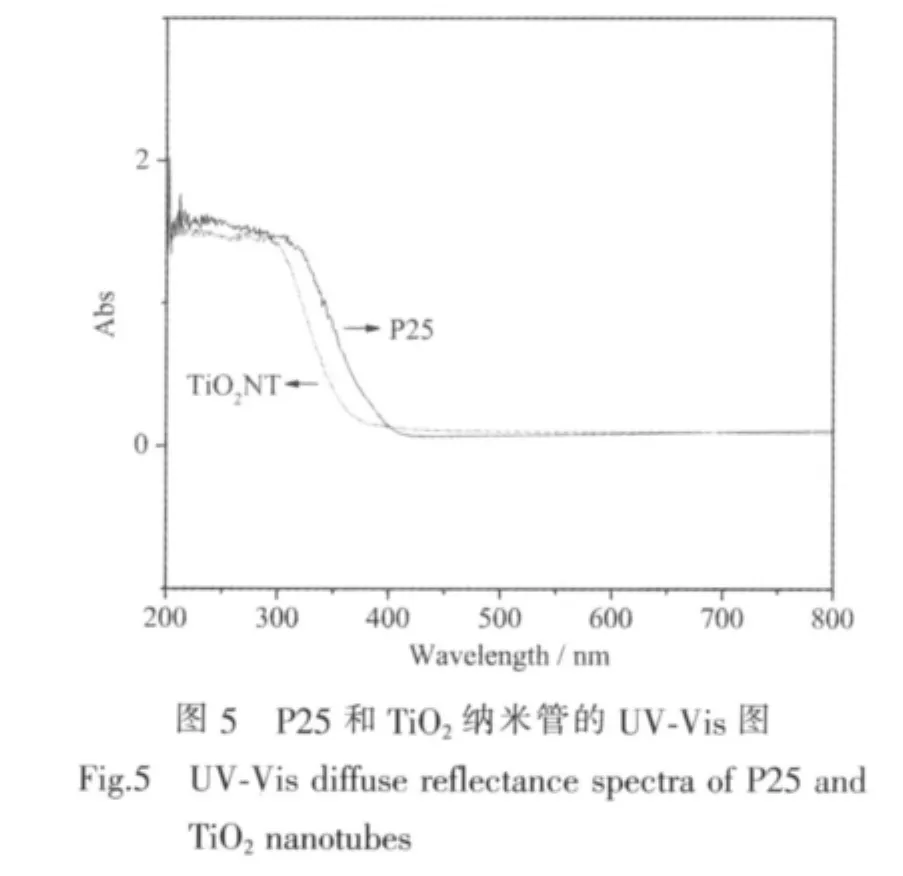
图5是P25和制备的二氧化钛纳米管样品的紫外-可见吸收光谱图,由图可知,与原料P25相比,纳米管的紫外吸收光谱发生了蓝移。图2所示的TiO2纳米管的SEM形貌观察表明,其平均管径约为10 nm,而原料P25的粒径为10~50 nm,表明由水热处理制备的TiO2纳米管的管壁是由很小的晶粒构成,这些晶粒的尺寸要明显小于原料P25(与XRD中衍射峰宽化的结果一致)。纳米粒子量子尺寸效应认为,小尺寸颗粒会使红外吸收带的精细结构消失,引起谱带宽化,吸收峰蓝移[22]。因此TiO2纳米管在紫外可见吸收光谱上表现为吸收边界相对于原料P25发生了蓝移。
3.4 光催化辅助电解水制氢
图6为不同阳极材料在有、无紫外光辐照下,阴极区氢气浓度与时间关系的对比图 (电解池电压为1.7 V),对图6进行线性拟合得到的斜率即为产氢速率R,结果列于表1。对单纯镍片(镍片未经修饰处理)电极,紫外光辐照对其产氢效率没有显著影响;在无光照情况下,纯Ni片作阳极与TiO2修饰阳极的产氢效率基本相同;当存在光照时,TiO2纳米管修饰阳极的电解水产氢速率比P25修饰阳极提高了20%,比单纯的Ni片作阳极提高了61%,从而实验初步验证了本文提出的光催化与电解对产氢的耦合作用的存在,并且纳米管结构的二氧化钛比颗粒状的P25具有更高的活性。
光解水与电解水的耦合作用如以下电极反应所示:

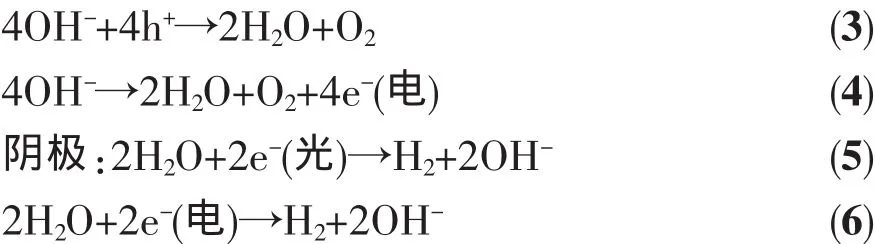
hⅤ、h+、e-(光)和 e-(电)分别代表光子、光生空穴、光生电子和直流电源贡献的电子。(2)、(3)和(5)是光催化过程,(4)和(6)是电解过程,在光催化辅助电解水中,同时发生了光催化和电解过程。
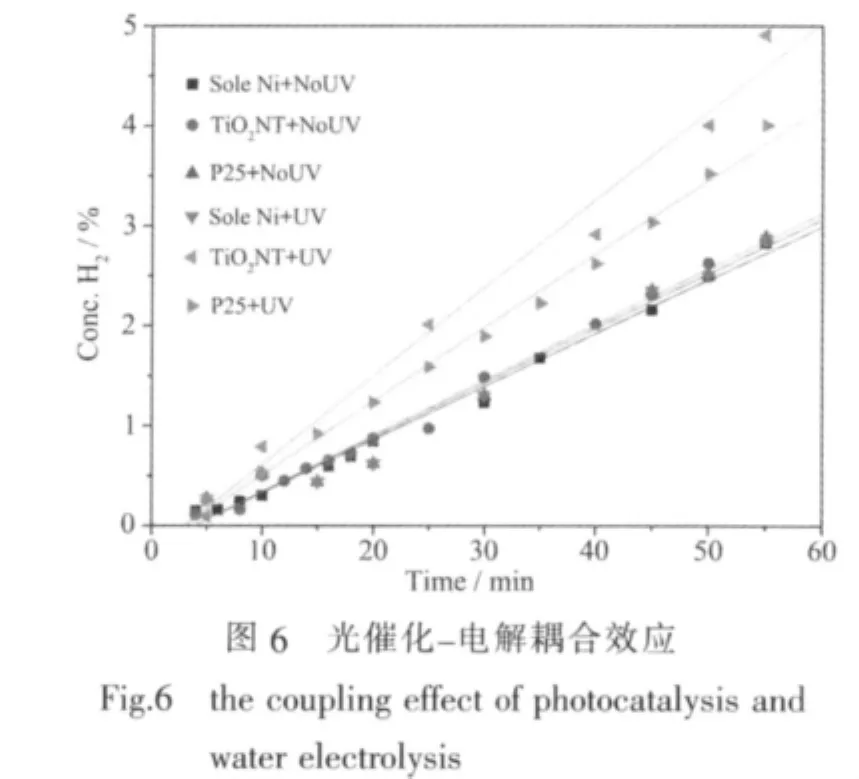
图7为以TiO2纳米管修饰镍片为阳极的电解水装置在单独光、电作用下和光电共同作用下的产氢情况。在没有紫外光辐照时,即为传统的电解水制氢过程,在本实验条件下,电解池电压低于1.6 V时,都没有观察到明显的产氢现象。单独紫外光照射(0 V+UV)时,甚至在施加直流电压低于1.6 V并同时有紫外光照射 (如图中列出的1 V+UV和1.5 V+UV)时,也都没有观察和检测到H2的产生,这与很多文献报道的光催化制氢和光电化学制氢的研究结果不同,可能是因为文献中使用具有很低析氢过电位、但价格昂贵和较难推广应用的贵金属Pt作阴极,或者是加入了牺牲剂(如乙二醇、Na2S),或者采用了抽真空的低压条件,另外文献报道的光电化学制氢体系中施加的偏压一般低于1 V,实际上没有电解水过程发生。Li[23]等报道了阳极氧化TiO2纳米管/Ti作光阳极,Pt/C作阴极,1 mol·L-1KOH作电解液,0.6 V 偏压下光电产氢速率为 0.178 mL·h-1·cm-2。张建灵[24]等报道了CdS/TiO2纳米管/Ti作光阳极,镍片作阴极,Na2S和Na2SO3为电子给体,1 V偏压下,光电产氢速率达到 0.245 mL·h-1·cm-2。而本实验采用的是以传统的碱性电解槽为基础,Ni-Cr合金作阴极,30%NaOH为电解质的工业化应用的电解水条件[25]。

表1 不同阳极材料的光催化辅助电解水产氢速率Table 1 Hydrogen production rate of WEAP with different anodes
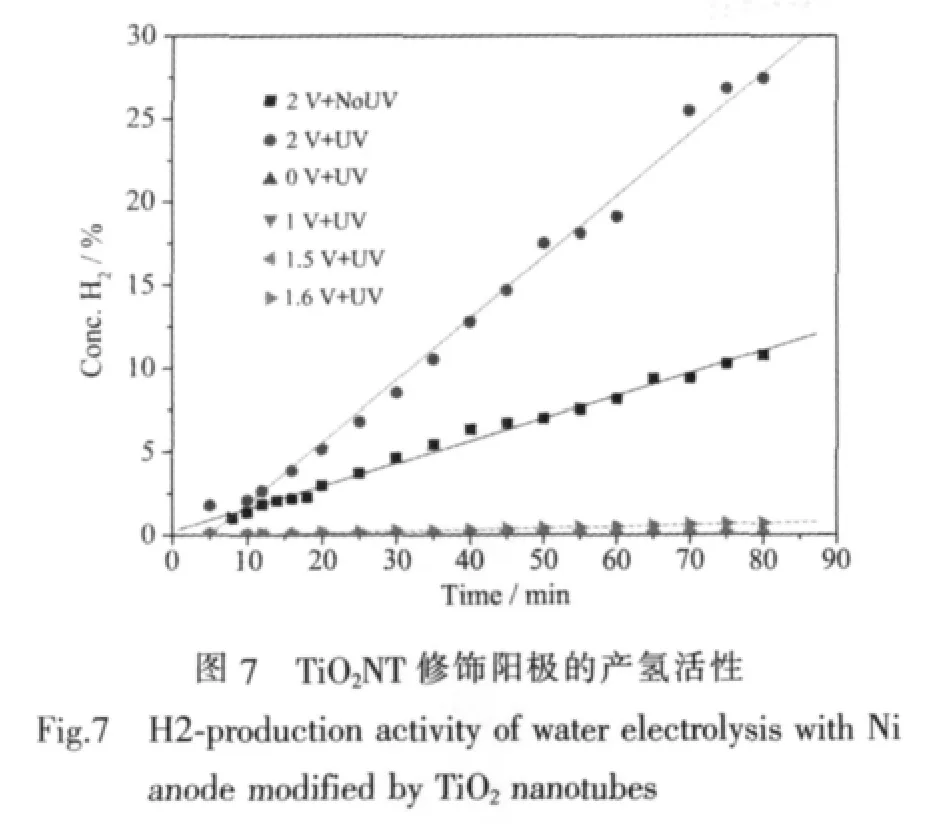
由图7的实验结果可知,在本实验条件下,只有当直流电压达到1.6 V时,才能克服理论水分解化学势、电极过电位和电解池阻抗,电解水产氢过程才能进行。只有在电解过程能够发生的前提下,用紫外光辐照TiO2光催化剂改性的阳极 (如图7中2 V+UV),产氢速率才会显著加快。表明光电耦合作用的基础是
电解水过程已经发生,在电解水的基础上耦合光催化过程构成的光催化辅助电解水的产氢效率远高于单独光、电作用时产氢量的简单加和,表现出明显的光电协同耦合产氢效应。说明只有在电解水产氢过程发生的情况下,光催化辅助的效果才能体现出来。
阳极涂覆的光催化剂在一定波长光线照射下能够产生电子空穴对,具有还原性的电子将H+还原为氢气,而具有氧化性的空穴则将OH-氧化为氧气,从而实现水的光解。然而,光生电子空穴对极易在短时间内复合,通常载流子间的复合是纳秒级快速过程,绝大多数光生电子和空穴还未来得及迁移到催化材料的表面参与氧化还原反应,就已经复合消耗了,导致单独光催化产H2效率极低。在本实验没有采用贵金属对光催化剂进行修饰、没有加入氢牺牲剂、常温常压和空气气氛等条件下,没有观察和检测到单独光催化产氢现象。在本文提出的光催化辅助电解水过程中,通过电解池的直流电压,将光生电子通过外电路及时转移到阴极,在阴极实现H+的还原产生氢气,阳极的空穴被电子给体OH-捕获,产生O2,促进了光生电子空穴的有效分离,因此将光解水制氢与电解水制氢有机地耦合在一起,产生了协同效应,显著地提高了产氢速率。
五结论
(1)采用水热法成功制备了平均外径为10 nm的TiO2纳米管光催化剂,并涂覆在电解池的阳极镍片上,形成了均匀的厚度约6~7 μm的光活性膜。
(2)制备的TiO2纳米管比颗粒状P25具有更高的光活性。
(3)光催化辅助电解水制氢实验表明,将TiO2纳米管光催化剂涂覆在电解池的阳极上,通过光对阳极的辐照,产氢速度比单纯的电解水过程提高了61%。初步验证了提出的光催化辅助电解水制氢的过程。
[1]Graf D,Monnerie N,Roeb M.Int.J.Hydrogen Energy,2009,33:4511-4519
[2]Clarke R E,Giddey S,Ciacchi F T.Int.J.Hydrogen Energy,2009,34:2531-2542
[3]Cherigui A N,Mahmah B,Harouadi F.Int.J.Hydrogen Energy.,2009,34:4934-4940
[4]NI Meng(倪萌),Leung M K H,Sumathy K.Energy Environmental Protection(Nengyuan Huanjing Baohu),2004,18(5):5-9
[5]Fujishima A,Honda K.Nature,1972,238:37-38
[6]WAN Bin(万斌),SHEN Jia-Nian(沈嘉年),CHEN Ming-Bo(陈鸣波),et al.Acta Chim.Sin.(Huaxue Xuebao),2008,66(11):1301-1306
[7]WANG Hou-Jin(王后锦),WU Xiao-Jing(吴晓婧),HUANG Lang-Huan(黄浪欢),et al.Chinese J.Catal.(Cuihua Xuebao),2011,32(4):637-642
[8]SUN Lan(孙岚),LI Jing(李静),WANG Cheng-Lin(王成林),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(2):334-338
[9]Wu N Q,Wang J,Tafen D N,et al.J.Am.Chem.Soc.,2010,132(19):6679-6685
[10]LIU Rong-Fang(刘蓉芳),CHENG Fang-Yi(程方益),TAO Zhan-Liang(陶 占 良),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(6):1021-1026
[11]QI Xiao-Wang(奚小网),HU Lin-Hua(胡林华),FANG Xia-Qin(方霞琴),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(7):1353-1357
[12]Lei Z B,You W S,Liu M Y,et al.Chem.Commun.,2003,17:2142-2143
[13]Lei Z B,Ma G J,Liu M Y,et al.J.Catal.,2006,237:322-329
[14]WEN Xin-Yu(文新宇),HUANG Zi-Yang(黄紫洋).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(6):1128-1132
[15]WEN Fu-Yu(温 福 宇),YANG Jin-Hui(杨 金 辉),LI Can(李灿),et al.Progress in Chemistry(Huaxue Jinzhan),2009,21(11):2285-2302
[16]TIAN Meng-Kui(田蒙奎),SHANGGUAN Wen-Feng(上官文峰),OUYANG Zi-Yuan(欧阳自远),et al.Journal of Functional Materials(Gongneng Cailiao),2005,36(10):1489-1492
[17]LI Qiong-Jiu(李琼玖),WANG Jian-Hua(王建华),LI De-Kuan(李德宽),etal.SinoGlobalEnergy(ZhongwaiNengyuan),2008,13(3):35-42
[18]HUANG Jin-Zhao(黄金昭),XU Zheng(徐征).Journal of Linyi University(Linyi Shifan Xueyuan Xuebao),2004,26(6):10-13
[19]Bach U,Lupo D,Comte P,et al.Nature,1998,395:583-585
[20]CAI Fang-Gong(蔡芳共),Yang Feng(杨峰),ZHAO Yong(赵勇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(3):504-508
[21]LI Hong-Yi(李洪义),WANG Jin-Shu(王金淑),CHEN Xin(陈欣),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26:217-222
[22]CUI Qiang(崔强),FENG Bo(冯波),CHEN Wei(陈伟),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,25:233-239
[23]Li Y K,Yu H M,Wei S,et al.Int.J.Hydrogen Energy,2011,36:14374-14380
[24]ZHANG Jian-Ling(张建灵),ZHANG Xing-Wang(张兴旺),LEI Le-Cheng(雷乐成).Chinese Science Bulletin(Kexue Tongbao),2008,12(53):1471-1473.
[25]WU Yu-Ping(吴玉萍),ZHOU Zhong-Hua(周忠华),HUANG Yue(黄悦),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,27(3):473-479
[26]Zeng K,Zhang D K.Prog.Energgy Combust Sci.,2010,36(3):307-326
[27]WANG Yi-zhong(王怡中),FU Yan(符雁),TANG Hong-xiao(汤鸿霄).Acta Scientiae Circumstantiate(Huanjing Kexue Xuebao),1999,19(1):65-69
[28]YE Zhao(叶钊),ZHANG Han-hui(张汉辉),PAN Hai-bo(潘海波).Spectroscopy and Spectral Analysis(Guangpuxue Yu Guangpu Fenxi),2002,22(6):928-931
[29]Mohapatra S K,Raja K S,Mahajan V K,et al.J.Phys.Chem.C.,2008,112:11007-11012
[30]Cho I S,Chen Z B,Forman A J,et al.Nano Lett.,2011,11:4978-4984
[31]SONG Gang-Xiang(宋刚祥).Thesis for the Master of Tianjing University(天津大学硕士论文).2008.
Nickel Anode Modified by TiO2Nanotubes and Photocatalytically Assisted Water Electrolysis for Hydrogen Generation
HE Hong-Bo CHANG Ming CHEN Ai-Ping*MA LeiDONG Hai-Jun LI Chun-Zhong
(Key Laboratory for Ultrane Materials of Ministry of Education,School of Materials Science and Engineering,East China University of Science and Technology,Shanghai 200237,China)
TiO2nanotubes(TiO2NT)were successfully synthesized by hydrothermal method.The TiO2nanotubes were coated onto Ni anodes for water electrolysis.A new hydrogen generation process,called as water electrolysis assisted by photocatalysis(WEAP),was proposed based on the coupling of water electrolysis with photocatalytic hydrogen evolution.The samples were characterized by X-ray diffraction(XRD),UV-Vis absorption spectroscope,field emission scanning electron microscope(FESEM),atomic force microscope(AFM)and WEAP measurement.The results showed that the rate of hydrogen generation increased by 61%in WEAP with the anode modified with TiO2NT in comparison with that in sole water electrolysis with unmodified Ni anode.
TiO2nanotubes;nickel anode modification;water electrolysis;photocatalysis;hydrogen generation
O614.41+1;TQ116.2+1
A
1001-4861(2012)10-2097-06
2012-01-09。收修改稿日期:2012-04-24。
国家自然科学基金(No.20925621)和上海市科委纳米专项(No.1052nm02400)资助项目。
*通讯联系人。E-mail:apchen@ecust.edu.cn,Tel:(021)64250996
猜你喜欢
陶瓷学报(2021年4期)2021-10-14
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
陶瓷学报(2019年5期)2019-01-12
分析化学(2018年12期)2018-01-22
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
化工管理(2017年25期)2017-03-05
中国塑料(2016年8期)2016-06-27
中国塑料(2016年12期)2016-06-15
应用化工(2014年11期)2014-08-16
